Infantile Refsum disease
| Infantile Refsum disease | |
|---|---|
| 'Other names: Infantile phytanic acid storage disease[1] | |
| Phytanic acid | |
Infantile Refsum disease (IRD) is a rare autosomal recessive[2] congenital peroxisomal biogenesis disorder within the Zellweger spectrum. These are disorders of the peroxisomes that are clinically similar to Zellweger syndrome and associated with mutations in the PEX family of genes.[3][4] IRD is associated with deficient phytanic acid catabolism, as is adult Refsum disease, but they are different disorders that should not be confused.[5]
Signs and symptoms
Infantile Refsum disease is one of three peroxisome biogenesis disorders which belong to the Zellweger spectrum of peroxisome biogenesis disorders (PBD-ZSD).[6] The other two disorders are Zellweger syndrome (ZS) and neonatal adrenoleukodystrophy (NALD).[7][8] Although they share a similar molecular basis for disease, Infantile Refsum disease is less severe than Zellweger syndrome.[9]
Infantile Refsum disease is a developmental brain disorder.[6] In addition, patients can show a reduction in central nervous system (CNS) myelin (particularly cerebral), which is referred to as (hypomyelination). Myelin is critical for normal CNS functions. Patients can also show postdevelopmental sensorineuronal degeneration that leads to a progressive loss of hearing and vision.[6]
Infantile Refsum disease can also affect the function of many other organ systems. Patients can show craniofacial abnormalities, hepatomegaly (enlarged liver), and progressive adrenal dysfunction.[6][9] Newborns may present with profound hypotonia (low muscle tone), and a poor ability to feed.[6][9] In some patients, a progressive leukodystrophy has been observed that has a variable age of onset.[9]
Molecular mechanism
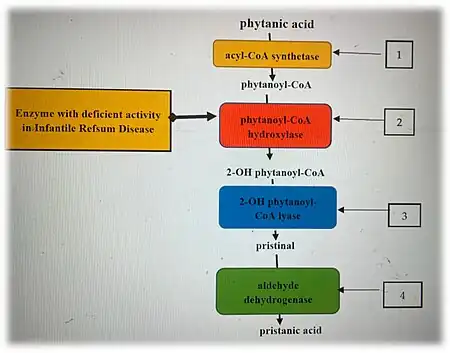
Infantile Refsum disease is an autosomal recessive disorder caused by mutations in genes that encode peroxins, proteins required for normal peroxisome assembly. Most commonly, patients have mutations in the PEX1, PEX3, PEX6, PEX12, and PEX26 genes.[1] In almost all cases, patients have mutations that inactivate or greatly reduce the activity of both the maternal and paternal copies of one these aforementioned PEX genes.
As a result of impaired peroxisome function, an individual's tissues and cells can accumulate very long chain fatty acids (VLCFA) and branched chain fatty acids (BCFA) that are normally degraded in peroxisomes. The accumulation of these lipids can impair the normal function of multiple organ systems, as discussed below. In addition, these individuals can show deficient levels of plasmalogens, ether-phospholipids that are especially important for brain, lung, and heart functions.
Diagnosis
In addition to genetic tests involving PEX genes,[11][12] biochemical tests have proven highly effective for the diagnosis of infantile Refsum disease and other peroxisomal disorders. Typically, IRD patients show elevated very long chain fatty acids in their blood plasma. Cultured primarily skin fibroblasts obtained from patients show elevated very long chain fatty acids, impaired very long chain fatty acid beta-oxidation, phytanic acid alpha-oxidation, pristanic acid alpha-oxidation, and plasmalogen biosynthesis.[6]
Management
Currently, there is no cure for infantile Refsum disease syndrome, nor is there a standard course of treatment. Infections should be guarded against to prevent such complications as pneumonia and respiratory distress. Other treatment is symptomatic and supportive. [9]
Prognosis
Patients show variable lifespans with some individuals surviving until adulthood and into old age.[9]
References
- 1 2 Online Mendelian Inheritance in Man (OMIM): Refsum Disease, Infantile form - 266510
- ↑ Choksi, V; Hoeffner, E; Karaarslan, E; Yalcinkaya, C; Cakirer, S (2003). "Infantile refsum disease: case report". AJNR. American Journal of Neuroradiology. 24 (10): 2082–4. PMC 8148918. PMID 14625237.
- ↑ Steinberg SJ, Raymond GV, Braverman NE, et al. (2020). Adam MP, Ardinger HH, Pagon RA, et al. (eds.). "Zellweger Spectrum Disorder". GeneReviews® [Internet]. University of Washington. PMID 20301621. NBK1448. Archived from the original on 2023-02-03. Retrieved 2023-08-28.
- ↑ Krause, C.; Rosewich, H.; Gärtner, J. (2009). "Rational diagnostic strategy for Zellweger syndrome spectrum patients". European Journal of Human Genetics. 17 (6): 741–8. doi:10.1038/ejhg.2008.252. PMC 2947092. PMID 19142205.
- ↑ Online Mendelian Inheritance in Man (OMIM): Refsum Disease, Classic - 266500
- 1 2 3 4 5 6 Steinberg, S.; Dodt, G.; Raymond, G.; Braverman, N.; Moser, A.; Moser, H. (2006). "Peroxisome biogenesis disorders". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1763 (12): 1733–48. doi:10.1016/j.bbamcr.2006.09.010. PMID 17055079.
- ↑ "GeneReviews: Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum". Archived from the original on 2007-05-10. Retrieved 2023-08-28.
- ↑ Krause, C.; Rosewich, H.; Thanos, M.; Gärtner, J. (2006). "Identification of novel mutations in PEX2, PEX6, PEX10, PEX12, and PEX13 in Zellweger spectrum patients". Human Mutation. 27 (11): 1157. doi:10.1002/humu.9462. PMID 17041890. S2CID 9905589.
- 1 2 3 4 5 6 Raymond, G. V.; Watkins, P.; Steinberg, S.; Powers, J. (2009). "Peroxisomal Disorders". Handbook of Neurochemistry and Molecular Neurobiology. pp. 631–670. doi:10.1007/978-0-387-30378-9_26. ISBN 978-0-387-30345-1.
- ↑ Slanina, Ana-Maria; Coman, Adorata-Elena; Anton-Păduraru, Dana-Teodora; Popa, Elena; Barbacariu, Carmen-Liliana; Novac, Otilia; Petroaie, Antoneta Dacia; Bacușcă, Agnes-Iacinta; Manole, Mihaela; Cosmescu, Adriana (9 March 2023). "PEX6 Mutation in a Child with Infantile Refsum Disease—A Case Report and Literature Review". Children. 10 (3): 530. doi:10.3390/children10030530. ISSN 2227-9067.
- ↑ Steinberg, S.; Chen, L.; Wei, L.; Moser, A.; Moser, H.; Cutting, G.; Braverman, N. (2004). "The PEX Gene Screen: molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum". Molecular Genetics and Metabolism. 83 (3): 252–263. doi:10.1016/j.ymgme.2004.08.008. PMID 15542397.
- ↑ Yik, W. Y.; Steinberg, S. J.; Moser, A. B.; Moser, H. W.; Hacia, J. G. (2009). "Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders". Human Mutation. 30 (3): E467–80. doi:10.1002/humu.20932. PMC 2649967. PMID 19105186.
Further reading
- Steinberg SJ, Raymond GV, Braverman NE, et al. (2020). Adam MP, Ardinger HH, Pagon RA, et al. (eds.). "Zellweger Spectrum Disorder". GeneReviews® [Internet]. University of Washington. PMID 20301621. NBK1448. Archived from the original on 2023-02-03. Retrieved 2023-08-28.
- OMIM entries on Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum 170993, 202370, 214100, 266510, 600279, 600414, 601498, 601758, 601789, 601791, 602136, 602859, 603164, 603360, 608666
External links
| Classification | |
|---|---|
| External resources |
|