Familial hypertriglyceridemia
| Familial hypertriglyceridemia | |
|---|---|
| Other names: Type IV familial dyslipidemia | |
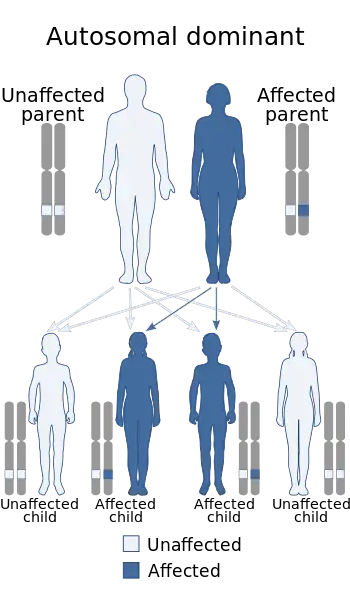 | |
| Familial hypertriglyceridemia is inherited in autosomal dominant manner | |
Familial hypertriglyceridemia (type IV familial dyslipidemia) is a genetic disorder characterized by the liver overproducing very-low-density lipoproteins (VLDL). As a result, an affected individual will have an excessive number of VLDL and triglycerides on a lipid profile. This genetic disorder usually follows an autosomal dominant inheritance pattern. The disorder presents clinically in patients with mild to moderate elevations in triglyceride levels. Familial hypertriglyceridemia is typically associated with other co-morbid conditions such as hypertension, obesity, and hyperglycemia. Individuals with the disorder are mostly heterozygous in an inactivating mutation of the gene encoding for lipoprotein lipase (LPL). This sole mutation can markedly elevate serum triglyceride levels. However, when combined with other medications or pathologies it can further elevate serum triglyceride levels to pathologic levels.[1] Substantial increases in serum triglyceride levels can lead to certain clinical signs and the development of acute pancreatitis.
Familial hypertriglyceridemia falls in the Fredrickson-Levy and Lee's (FLL) phenotypes. The phenotypes include types I, IIa, IIb, III, IV, and V dyslipidemias. Familial hypertriglyceridemia is considered a type IV familial dyslipidemia it is distinguished from other dyslipidemias based on the individual's lipid profile. Familial hypertriglyceridemia separates itself from other dyslipidemias with significantly high triglycerides and low HDL levels. It is important to recognize that co-morbid conditions that often concomitantly exist with the disorder can further alter the lipid panel.[2]
Signs and symptoms
There is usually no presentation in Familial hypertriglyceridemia[3]
Cause
Familial hypertriglyceridemia is considered to be inherited in an autosomal dominant manner. However, it is important to recognize that most cases have a polygenic inheritance distancing themselves from traditional Mendelian inheritance patterns.[4] One of the most common mutations implicated in the development of familial hypertriglyceridemia is a heterozygous inactivating mutation of the LPL gene. Inactivation of this gene leads to an individual's inability to hydrolyze the triglycerides within the VLDL core. This inactivation of function leads to a considerable accumulation of triglycerides and VLDL in the bloodstream, which then contributes to several avenues of pathology. Individuals with insulin resistance can have even further elevated levels of hypertriglyceridemia due to the fact that insulin is a potent activator of LPL. Therefore, an individual who is resistant to the bioactivity of insulin will have decreased LPL activity and will therefore lead to further hypertriglyceridemia, helping push serum triglycerides to pathologic levels. Beyond the classic understanding of single-gene mutation leading to disease, hypertriglyceridemia is also linked to several different genetic loci permitting additional aberrant changes to other lipid levels in the body.[5]
Pathophysiology
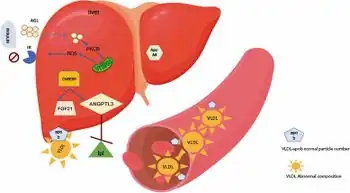
Inactivity of lipoprotein lipase (LPL) plays the predominant role in the development of familial hypertriglyceridemia. LPL plays a role in the metabolism of triglycerides within VLDL molecules. Inactivation mutations in LPL will create an environment with an increased concentration of VLDL molecules and therefore, triglycerides. The elevation of baseline triglyceride levels begins the cascade into other pathologies.[2]
The most common acute manifestation of hypertriglyceridemia is the occurrence of pancreatitis. Pancreatitis is caused by the premature activation of exocrine pancreatic enzymes. Secreted zymogens are cleaved to active trypsin and play a central role in digestion of food in the duodenum. If there is premature activation of trypsin within the pancreatic tissues, there is an induction of autodigestion of local tissue which leads to the initial presentation of pancreatitis. Autodigestion of local tissues also leads to disruptions in pancreatic microvascular tissue which can cause an ischemia-reperfusion event at the pancreatic level. There are other varying secondary causes of pancreatitis that can further contribute to the primary scenario of pancreatitis related to familial hypertriglyceridemia.[7][8]
Diagnosis
The evaluation of Familial hypertriglyceridemia can be done via a fasting lipid panel screening[3]
Treatment
Treatment for familial hypertriglyceridemia should focus primarily on reducing serum triglyceride levels. If an individual has co-morbid conditions, ensuring that they are adequately addressed will aid in obtaining a more normal baseline lipid panel. Current guidelines suggest that when evaluating individuals with familial hypertriglyceridemia there should be special attention paid to their risk of developing cardiovascular disease in individuals with mild to moderate hypertriglyceridemia. Individuals with severe hypertriglyceridemia should be promptly evaluated for the possibility of developing pancreatitis.[7] The initial treatment for severe hypertriglyceridemia consists of beginning an individual on fibrate therapy in an attempt to normalize triglyceride levels. Fibrates such as fenofibrate or gemfibrozil are considered first-line therapy for the disease. Adjunctive niacin therapy can be used for individuals who are unable to decrease triglyceride levels through fibrate monotherapy. Niacin is especially useful for individuals who have a high risk of getting pancreatitis. Fish oil supplement can also be used as it has been shown to incur a significant reduction to both triglyceride and VLDL levels.[9] If properly managed, individuals with familial hypertriglyceridemia have a fairly good prognosis. If therapy is successful, these individuals do not have uncontrolled severe triglycerides and VLDL. It is important to educate individuals on possible secondary causes of elevated lipid profiles. Proper management of the secondary causes provides a good prognosis for overall individual health.
Epidemiology
Familial hypertriglyceridemia can follow an autosomal dominant monogenic inheritance pattern. The frequency of heterozygous carriers of certain pathologic mutations in the LPL gene can range from 0.06% to 20%. It is important to note that dissimilar mutations can confer varying degrees of underlying pathology. However, most cases of familial hypertriglyceridemia follow a polygenic inheritance pattern involving mutations in multiple genetic foci.[10][11]
See also
References
- ↑ Ripatti P, Rämö JT, Mars NJ, Fu Y, Lin J, Söderlund S, et al. (April 2020). "Polygenic Hyperlipidemias and Coronary Artery Disease Risk". Circulation: Genomic and Precision Medicine. 13 (2): e002725. doi:10.1161/CIRCGEN.119.002725. PMC 7176338. PMID 32154731.
- 1 2 Quispe R, Hendrani A, Baradaran-Noveiry B, Martin S, Brown E, Kulkarni K, et al. (2019). "Characterization of lipoprotein profiles in patients with hypertriglyceridemic Fredrickson-Levy and Lees dyslipidemia phenotypes: the Very Large Database of Lipids Studies 6 and 7". Archives of Medical Science. 15 (5): 1195–1202. doi:10.5114/aoms.2019.87207. PMC 6764300. PMID 31572464. S2CID 203620865.
- 1 2 Goyal, Amandeep; Cusick, Austin S.; Reilly, Elizabeth (2023). "Familial Hypertriglyceridemia". StatPearls. StatPearls Publishing. Archived from the original on 6 February 2023. Retrieved 12 May 2023.
- ↑ Hegele RA, Ban MR, Hsueh N, Kennedy BA, Cao H, Zou GY, et al. (1 November 2009). "A polygenic basis for four classical Fredrickson hyperlipoproteinemia phenotypes that are characterized by hypertriglyceridemia". Human Molecular Genetics. 18 (21): 4189–4194. doi:10.1093/hmg/ddp361. PMC 2758142. PMID 19656773.
- ↑ Johansen CT, Wang J, Lanktree MB, McIntyre AD, Ban MR, Martins RA, et al. (August 2011). "An Increased Burden of Common and Rare Lipid-Associated Risk Alleles Contributes to the Phenotypic Spectrum of Hypertriglyceridemia". Arteriosclerosis, Thrombosis, and Vascular Biology. 31 (8): 1916–1926. doi:10.1161/ATVBAHA.111.226365. PMC 3562702. PMID 21597005. S2CID 7920385.
- ↑ Cruz-Bautista, Ivette; Huerta-Chagoya, Alicia; Moreno-Macías, Hortensia; Rodríguez-Guillén, Rosario; Ordóñez-Sánchez, María Luisa; Segura-Kato, Yayoi; Mehta, Roopa; Almeda-Valdés, Paloma; Gómez-Munguía, Lizeth; Ruiz-De Chávez, Ximena; Rosas-Flota, Ximena; Andrade-Amado, Arali; Bernal-Barroeta, Bárbara; López-Carrasco, María Guadalupe; Guillén-Pineda, Luz Elizabeth; López-Estrada, Angelina; Elías-López, Daniel; Martagón-Rosado, Alexandro J.; Gómez-Velasco, Donají; Lam-Chung, Cesar Ernesto; Bello-Chavolla, Omar Yaxmehen; Del Razo-Olvera, Fabiola; Cetina-Pérez, Lucely D.; Acosta-Rodríguez, José Luis; Tusié-Luna, María Teresa; Aguilar-Salinas, Carlos A. (15 February 2021). "Familial hypertriglyceridemia: an entity with distinguishable features from other causes of hypertriglyceridemia". Lipids in Health and Disease. 20 (1): 14. doi:10.1186/s12944-021-01436-6. ISSN 1476-511X. Retrieved 12 May 2023.
- 1 2 Berglund L, Brunzell JD, Goldberg AC, Goldberg IJ, Sacks F, Murad MH, et al. (1 September 2012). "Evaluation and Treatment of Hypertriglyceridemia: An Endocrine Society Clinical Practice Guideline". The Journal of Clinical Endocrinology & Metabolism. 97 (9): 2969–2989. doi:10.1210/jc.2011-3213. PMC 3431581. PMID 22962670.
- ↑ "Corrigenda". The Journal of Clinical Endocrinology & Metabolism. 100 (12): 4685. 1 December 2015. doi:10.1210/jc.2015-3649. PMC 5399508. PMID 26562756.
- ↑ Pradhan A, Bhandari M, Vishwakarma P, Sethi R (March 2020). "Triglycerides and Cardiovascular Outcomes—Can We REDUCE-IT ?". International Journal of Angiology. 29 (1): 002–011. doi:10.1055/s-0040-1701639. PMC 7054063. PMID 32132810.
- ↑ Triglyceride Coronary Disease Genetics Consortium Emerging Risk Factors Collaboration, Sarwar N, Sandhu MS, Ricketts SL, Butterworth AS, Di Angelantonio E, et al. (May 2010). "Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies". The Lancet. 375 (9726): 1634–1639. doi:10.1016/S0140-6736(10)60545-4. PMC 2867029. PMID 20452521.
- ↑ "Department of Error". The Lancet. 376 (9735): 90. July 2010. doi:10.1016/S0140-6736(10)61075-6. PMC 3081093.
External links
| Classification |
|---|