Chylomicron retention disease
| Chylomicron retention disease | |
|---|---|
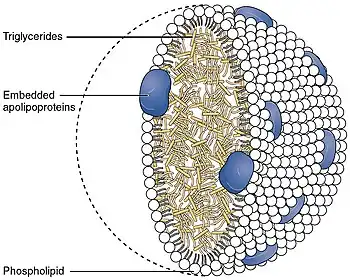 | |
| 2512 Chylomicrons Contain Triglycerides Cholesterol Molecules and Other Lipids | |
| Frequency | Lua error in Module:PrevalenceData at line 5: attempt to index field 'wikibase' (a nil value). |
Chylomicron retention disease is a disorder of fat absorption.[1] It is associated with SAR1B.[2] Mutations in SAR1B prevent the release of chylomicrons in the circulation which leads to nutritional and developmental problems.[3] It is a rare autosomal recessive disorder with around 40 cases reported worldwide. Since the disease allele is recessive, parents usually do not show symptoms.[3]
Without functional chylomicrons, certain fat-soluble vitamins such as vitamin D and vitamin E cannot be absorbed. Chylomicrons have a crucial role in fat absorption and transport, thus a deficiency in chylomicron functioning reduces available levels of dietary fats and fat-soluble vitamins.[3]
Signs and symptoms
Physical symptoms of CMRD involving development and function of the gastrointestinal tract and nervous system typically manifest between infancy and adolescence. The symptoms of CmRD are similar to the physical symptoms of malnutrition, as the disease arises due to the poor absorption of lipids and fat-soluble nutrients such as vitamin E. For this reason, the disease is likely to be under-diagnosed by physicians . Fat-soluble nutrients are essential for growth, development, and normal bodily function. Vitamin E deficiency is especially serious, as the vitamin is necessary for proper neurological function and development. Without Vitamin E, neurons cannot operate correctly and signals from the brain are weakened. This leads to reduced muscle development and reduced muscle contraction.
Symptoms that manifest in the GI tract are likely to be a consequence of both reduced absorption of fats and physiological stress imposed on enterocytes that can not shuttle fats into circulation . Additional symptoms that occur throughout the body can be attributed to the lack of sufficient lipid sources.[4]
- Chronic Malabsorptive Diarrhea- Diarrhea that results from the poor absorption of fats
- Steatorrhea- Abnormal stools, often foul smelling, due to the increased presence of undigested fats
- Vomiting
- Vitamin E Deficiency- Low levels of Vitamin E due to the malabsorption of fats in the diet, causes poor brain, muscle, and eye development.
- Cardiomyopathy- A class of disease that affects heart muscle, causing shortness of breath, tiredness, and swelling of the legs
- Slowed Growth
- Failure to Thrive- Insufficient weight gain, or drastic levels of weight loss in children
- Hypocholesterolemia- Low blood cholesterol levels
- Hepatic Steatosis (Fatty Liver)- Excessive fat buildup in the liver, a result of the abnormal lipid panels of CMRD patients
- Hyporeflexia- Absent or low levels of muscle reflexes
- Amyotrophy- Muscle tissue “wasting,” the loss of muscle tissue
Genetics
The Sar1B GTPase is an enzyme located in epithelial cells of the gastrointestinal tract. These proteins are critical for release of chylomicrons in the body.[5]
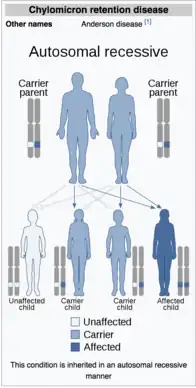
Chylomicron retention disease is an autosomal homozygous recessive disorder arising from mutations in the gene encoding the Sar1B GTPase. The Sar1B gene is located at position 5q31.1 in the fifth chromosome and is composed of eight exons. Alternative splicing of the second exon results into two different splice isoforms for the Sar1B transcript RNA. In CMRD, a mutation of this genomic sequence affects the Sar1B enzyme's ability to interact with Guanine Exchange Factors (GEFs) and GTP-Activating Proteins (GAPs). The mutation of exon 6 of the sequence can eliminate the critical chain that is responsible for recognizing guanine. This strips the GTPase of its capability to hydrolyze GTP, its hallmark trait. This overall affects the ability of Sar1B GTPase to control chylomicron release. A third mutant allele containing a missense mutation has also been reported to cause CMRD.[6] All three of these alleles display recessive inheritance, suggesting that they loss-of-function mutations cause the symptoms of CMRD.
Physiology
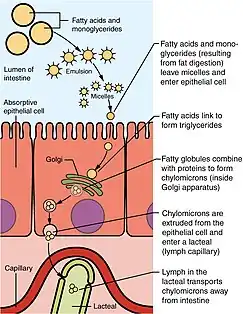
During digestion, fats, or triglycerides(TGs), are enzymatically catabolized by lipases into two fatty acids and a monoglyceride molecule. Those components are then transported across the enterocyte membrane as micelles and reformed into triglycerides once across the membrane.[7]
Once transported to the ER the triglycerides are incorporated into pre-chylomicrons which are made up of TGs, cholesterol, and phospholipids. The pre-chylomicrons are then packaged into PCTV in order to be transported to the Golgi apparatus for additional maturation prior to exocytosis into the lymphatic system.[8] From the lymphatic system, they enter general circulation, where they are produced in various forms that can be absorbed by bodily tissues and metabolized or stored by adipose tissue. Before the PCTV leaves the ER, it is incorporated into a COPII coatomer of five proteins. The PCTV undergoes a similar mechanism for budding as normal COPII transport vesicles.[8] Though PCTV does not require COPII coatomer proteins for budding from the ER, association with the coatomer is necessary for docking and fusion with the cis-golgi network.[8] In chylomicron retention disease, the PCTV vesicles are competent for budding from the ER membrane but are defective for fusion with the cis-golgi body.
Sar1B is a GTPase and one of the five proteins of the COPll coatomer. A mutation in the sar1B gene and subsequently the sar1B protein are the common genetic origins of chylomicron retention disorder. Without the fully functional sar1B protein, the COPll coatomer proteins engulf pre-chylomicrons exiting the ER but are unable to disassemble upon arrival at the cis-Golgi, preventing membrane fusion with this organelle.[9]
Diagnosis
There is no medical consensus on methodology of diagnosis for CMRD itself. There are, however, protocols used to diagnose the family of genetic disorders to which CMRD belongs. Assessment of hypobetalipoproteinemia relies chiefly on blood lipid analysis following a 12-hr fasting period. Lipids analyzed are LDL (low-density lipoproteins), triglyceride, and apolipoprotein B levels. A patient could be diagnosed with CMRD should they lack sufficient apolipoprotein B levels in the blood. Furthermore, a minimally invasive endoscopic procedure can be used to examine the bowel. A pale intestine can also be indicative of CMRD.[5]
Because patient outcomes rely on early diagnosis, it is recommended that candidates for the disorder should receive lipid panel testing prior to 6 months of age. In patients with only CMRD, lipid panels are expected to display normal triglyceride levels, but LDL and HDL levels may >50% below normal range. The test should also reveal low levels of Vitamin E and heightened levels of creatine kinase in the blood.[10]
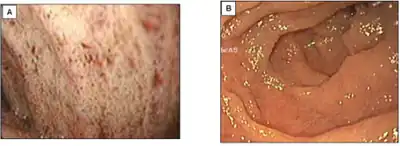 a)Endoscopy shows white duodenal mucosa (affected subject)
a)Endoscopy shows white duodenal mucosa (affected subject)
b) normal subject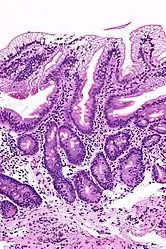 Abetalipoproteinemia - high mag
Abetalipoproteinemia - high mag
Treatment
It is recommended that patients with CMRD follow a strict low-fat diet in addition to fat-soluble vitamin supplementation. The fat soluble vitamins are A,D,E, and K. A combination of vitamin A and vitamin E are effective for combating ophthalmologic complications. When vitamin D is administered early, it aids in preventing osteopenia.[10] People with CMRD are at an increased risk for essential fatty acid deficiency, so dietary counseling is required in order to maintain the low-fat diet, while attaining sufficient caloric intake and essential fatty acid intake.[10]
Proposed Treatment Plan
Early diagnosis is important for improving patient outcomes. Patients with delayed diagnoses experienced decreased growth compared to those diagnosed earlier in life. Long-term treatment plans center around dietary management, but because long term results have not been documented due to a lack of thorough research, careful monitoring of the disease is required. Yearly check-ups are recommended to track the growth of children affected by the disease.
Evaluations tracking liver function that involve the use of ultrasounds to monitor liver growth, are recommended to be administered every three years. At about ten years of age (pre-puberty), neurological and ophthalmological exams may be required every three years to track muscle and eye activity/strength. In adulthood, past eighteen years of age, echocardiograms are recommended to track heart activity. Thorough and vigorous testing warrant themselves to the treatment of a disease of which we know so little.[11]
| Early Follow-Up(Annual) | |
|---|---|
| Clinical | |
| Anthropometry | Weight and height to draw growth curve |
| Digestive | Appetite, diarrhea, abdominal distension, vomiting, hepatic size? |
| Neurological | Developmental retardation, areflexia, ataxia, dysarthria, deep proprioception loss, muscular weakness or pain, cramps? |
| Dietary counseling | Sufficient caloric intake, low fat diet (fat <30% total energy), EFA supplementation? |
| Biological | |
| Lipids | Total and LDL cholesterol, HDL-C, TG |
| Hepatic | AST, ALT, GGT, total bilirubin, alkaline phosphatase? |
| Vitamins | Plasma levels of vitamins A, D, E and K or INR (vit K deficiency) |
| Essential Fatty Acids | Deficiency induced by low fat diet? |
| Blood cell count | Anemia? |
| Delayed Follow-Up (every 3 years) | |
| After the age of 10 years | |
| Hepatic | Ultrasonography (steatosis, portal hypertension, yearly), Elastometry Fibroscan®? (further studies are needed) |
| Neurological exam | Clinical, creatine kinase, electromyography |
| Ophthalmologic exam | Fundus, color vision, visual evoked potentials, electroretinography |
| Total body composition | Bone mineral content for whole body |
| Adult age | |
| Echocardiography | Ejection fraction |
Prognosis
As of March 2020, only 50 cases of CMRD have been documented in the medical literature.[4] This small number speaks to the rarity of the disease as well as the lack of thorough research and documentation. As a result, the full course of the disease, life expectancy, and mortality are also poorly documented.
Clinical manifestation of CMRD symptoms begin during infancy and early childhood but may go undetected due to the non-specific symptoms associated with the disease. Many of these symptoms can be attributed to malnutrition and nonspecific postnatal diarrhea, confounding early diagnosis. Careful regulation of diet and nutrition are required for management of CMRD since the disease results from the poor absorption of nutrients from food.
History
Chylomicron Retention Disease, also called Anderson's disease, is an autosomal recessive lipid malabsorption syndrome characterized by abnormally low amounts of cholesterol in the blood. This disease most frequently is diagnosed in infants. Charlotte Anderson first published a description of the disorder in 1961, where she observed a seven month old girl who developed intestinal mucosa filled with fat droplets. In 2003, Jones and colleagues identified mutations in the SAR1B gene, which transcripts the SAR1B protein involved in COPII transport and proposed this was the molecular defect of the disorder.[12] To present day, 17 mutations of the SAR1B gene have been discovered. This disease is rare, with only 50 cases diagnosed worldwide.
References
- ↑ Roy CC, Levy E, Green PH, et al. (February 1987). "Malabsorption, hypocholesterolemia, and fat-filled enterocytes with increased intestinal apoprotein B. Chylomicron retention disease". Gastroenterology. 92 (2): 390–9. doi:10.1016/0016-5085(87)90133-8. PMID 3792776.
- ↑ Jones B, Jones EL, Bonney SA, et al. (May 2003). "Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders". Nat. Genet. 34 (1): 29–31. doi:10.1038/ng1145. PMID 12692552. S2CID 10543077.
- 1 2 3 "Archive copy". Archived from the original on 2020-08-07. Retrieved 2021-02-05.
{{cite web}}: CS1 maint: archived copy as title (link) - 1 2 Reference, Genetics Home. "Chylomicron retention disease". Genetics Home Reference. Archived from the original on 2020-06-16. Retrieved 2020-05-04.
- 1 2 "Orphanet: Hypobetalipoproteinemia". www.orpha.net. Archived from the original on 2005-11-13. Retrieved 2020-05-07.
- ↑ Simone, Maria Luisa; Rabacchi, Claudio; Kuloglu, Zarife; Kansu, Aydan; Ensari, Arzu; Demir, Arzu Meltem; Hizal, Gulin; Di Leo, Enza; Bertolini, Stefano; Calandra, Sebastiano; Tarugi, Patrizia (July 2019). "Novel mutations of SAR1B gene in four children with chylomicron retention disease". Journal of Clinical Lipidology. 13 (4): 554–562. doi:10.1016/j.jacl.2019.05.013. ISSN 1933-2874. PMID 31253576.
- ↑ "Archive copy". Archived from the original on 2020-10-27. Retrieved 2021-02-05.
{{cite web}}: CS1 maint: archived copy as title (link) - 1 2 3 (https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-6-1 Archived 2021-01-15 at the Wayback Machine)
- ↑ Georges, Amandine; Bonneau, Jessica; Bonnefont-Rousselot, Dominique; Champigneulle, Jacqueline; Rabès, Jean P.; Abifadel, Marianne; Aparicio, Thomas; Guenedet, Jean C.; Bruckert, Eric; Boileau, Catherine; Morali, Alain; Varret, Mathilde; Aggerbeck, Lawrence P.; Samson-Bouma, Marie E. (2011). "Molecular analysis and intestinal expression of SAR1 genes and proteins in Anderson's disease (Chylomicron retention disease)". Orphanet Journal of Rare Diseases. 6: 1. doi:10.1186/1750-1172-6-1. PMID 21235735. S2CID 927898. Archived from the original on 2021-01-15. Retrieved 2021-02-05.
- 1 2 3 Peretti, Noel; Sassolas, Agnès; Roy, Claude C.; Deslandres, Colette; Charcosset, Mathilde; Castagnetti, Justine; Pugnet-Chardon, Laurence; Moulin, Philippe; Labarge, Sylvie; Bouthillier, Lise; Lachaux, Alain (2010-09-29). "Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers". Orphanet Journal of Rare Diseases. 5 (1): 24. doi:10.1186/1750-1172-5-24. ISSN 1750-1172. PMC 2956717. PMID 20920215.
- ↑ Peretti, Noel; Sassolas, Agnès; Roy, Claude C.; Deslandres, Colette; Charcosset, Mathilde; Castagnetti, Justine; Pugnet-Chardon, Laurence; Moulin, Philippe; Labarge, Sylvie; Bouthillier, Lise; Lachaux, Alain (2010-09-29). "Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers". Orphanet Journal of Rare Diseases. 5 (1): 24. doi:10.1186/1750-1172-5-24. ISSN 1750-1172. PMC 2956717. PMID 20920215.
- ↑ Jones, Bethan; Jones, Emma L.; Bonney, Stephanie A.; Patel, Hetal N.; Mensenkamp, Arjen R.; Eichenbaum-Voline, Sophie; Rudling, Mats; Myrdal, Urban; Annesi, Grazia; Naik, Sandhia; Meadows, Nigel; Quattrone, Aldo; Islam, Suhail A.; Naoumova, Rossitza P.; Angelin, Bo; Infante, Recaredo; Levy, Emile; Roy, Claude C.; Freemont, Paul S.; Scott, James; Shoulders, Carol C. (2003). "Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders". Nature Genetics. 34 (1): 29–31. doi:10.1038/ng1145. PMID 12692552. S2CID 10543077. Archived from the original on 2021-11-01. Retrieved 2021-02-05.
| Classification | |
|---|---|
| External resources |
|