Zaspopathy
| Zaspopathy | |
|---|---|
| Other names | Late-onset distal myopathy, Markesbery-Griggs type |
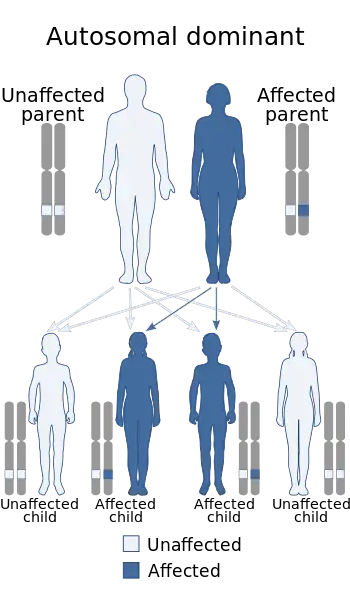 | |
| Zaspopathy has an autosomal dominant pattern of inheritance. | |
Zaspopathy,[1] also called ZASP-related myofibril myopathy,[2] is a novel autosomal dominant[3] form of progressive muscular dystrophy, first described in 2005.
Cause
The disease encompasses multiple forms of both distal and proximal myopathies, and is caused by mutations in the gene referred to as ZASP.[3]
Pathophysiology
The ZASP gene is located at chromosome 10, and encodes also-called Z-disk-associated protein.Mutation in this protein causes disintegration of the Z-disk of contractile elements (myofibrils) in muscle cells.
Mutations of several other Z-disk related proteins, such as desmin, alfa-B-crystallin and myotilin can cause disorders similar to zaspopathy.
Diagnosis
Treatment
References
- ↑ Griggs R, Vihola A, Hackman P, Talvinen K, Haravuori H, Faulkner G, Eymard B, Richard I, Selcen D, Engel A, Carpen O, Udd B (Jun 2007). "Zaspopathy in a large classic late-onset distal myopathy family" (Free full text). Brain : A Journal of Neurology. 130 (Pt 6): 1477–1484. doi:10.1093/brain/awm006. PMID 17337483.
- ↑ Online Mendelian Inheritance in Man (OMIM): 609452
- 1 2 Selcen D, Engel AG (Feb 2005). "Mutations in ZASP define a novel form of muscular dystrophy in humans". Annals of Neurology. 57 (2): 269–276. doi:10.1002/ana.20376. PMID 15668942. S2CID 25733755. Archived from the original on 2012-12-17.
External links
This article is issued from Offline. The text is licensed under Creative Commons - Attribution - Sharealike. Additional terms may apply for the media files.