Hypokalemic periodic paralysis
| Hypokalemic periodic paralysis | |
|---|---|
| Other names: Familial hypokalemic periodic paralysis (FHPP)[1] | |
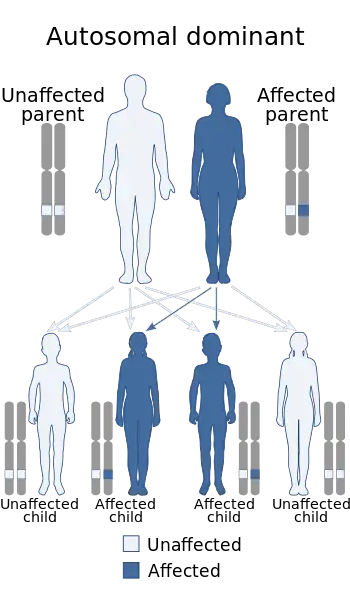 | |
| This condition is inherited in an autosomal dominant manner | |
| Specialty | Neurology |
| Symptoms | Episodes of severe muscle weakness[2] |
| Complications | Aspiration pneumonia, heart arrhythmias[2] |
| Usual onset | 10 to 30 years old[2] |
| Types | Familial, acquired[2] |
| Causes | Genetics, hyperthyroidism[2] |
| Differential diagnosis | Renal tubular acidosis, other causes of low potassium, thyrotoxic periodic paralysis, hyperkalemic periodic paralysis, myasthenia gravis[2] |
| Prevention | Avoiding triggers, potassium supplementation, acetazolamide, potassium-sparing diuretics[2] |
| Treatment | Potassium chloride by mouth[2] |
| Frequency | Rare[2] |
Hypokalemic periodic paralysis (hypoKPP) is a genetic condition characterized by episodes of severe muscle weakness.[2] While the underlying cause is present at birth, attacks do not generally begin until the age of 10 to 30.[2] Before the onset of weakness, tiredness, tingling, or behavioral changes may occur.[2] Onset is over minutes to hours and symptoms may last hours to days.[2][3] The frequency of attacks is variable.[2] Complications may include aspiration pneumonia, heart arrhythmias, and long term muscle damage.[2]
Most cases are due to a mutation in the CACNA1S gene and are autosomal dominant.[2] This mutation may be inherited from a parent or occur during early development.[2] Other cases may arise due to high thyroid, which is known as thyrotoxic periodic paralysis.[2] Attacks may be triggered following strenuous exercise or by a high carbohydrate diet, salt, alcohol, cold, psychological stress, or corticosteroids.[2] The underlying mechanism involves a defect in an ion channel which results in periods of low blood potassium.[2] The diagnosis is suspected based on symptoms and either a family history or low potassium during an attack.[2]
Efforts to prevent attacks include avoiding triggers, potassium supplementation, acetazolamide, and potassium-sparing diuretics.[2] Attacks are treated by giving potassium chloride at doses of 0.5 to 1 mEq/kg by mouth.[2] Further doses at 0.3 mEq/kg may be given ever 30 minutes if the symptoms do not improve to a maximum of 200 mEq per 24 hours.[2] Intravenous potassium may given, preferably in mannitol, for those who cannot swallow or have a heart arrhythmia.[2]
HypoKPP is rare, affecting about 1 in 100,000 people.[2] Women are less seriously affected than men.[2] Cases due to high thyroid occur more commonly in Asians and Latinx.[3] It was first described in 1727 by Musgrave.[2]
Signs and symptoms
Hypokalemic periodic paralysis is a condition that causes episodes of extreme muscle weakness typically beginning in childhood or adolescence. Most often, these episodes involve a temporary inability to move muscles in the arms and legs. Attacks cause severe weakness or paralysis that usually lasts from hours to days. Some people may have episodes almost every day, while others experience them weekly, monthly, or only rarely. Attacks can occur without warning or can be triggered by factors such as rest after exercise, a viral illness, or certain medications. Often, a large, carbohydrate-rich meal or vigorous exercise in the evening can trigger an attack upon waking the following morning. Although affected individuals usually regain their muscle strength between attacks, some develop persistent muscle weakness later in life.[4]
Genetics
Mutations in the following genes can cause hypokalemic periodic paralysis:
| Type | OMIM | Gene | Locus |
|---|---|---|---|
| HOKPP1 | 170400 | CACNA1S (a voltage-gated calcium channel Cav1.1 found in the transverse tubules of skeletal muscle cells) | 1q32 |
| HOKPP2 | 613345 | SCN4A (a voltage-gated sodium channel Nav1.4 found at the neuromuscular junction) | 17q23.1-q25.3 |
| 170390 | KCNJ2 (an inward-rectifier potassium channel Kir2.1) | 17q24.3 |
An association with KCNE3 (voltage-gated potassium channel) has also been described, but is currently disputed,[5] and excluded from the disease definition in OMIM.[6]
Action potentials from the central nervous system cause end-plate potentials at the NMJ which causes sodium ions to enter and depolarise the muscle cells. This depolarisation propagates to the T-tubules where it triggers the entry of calcium ions via Cav1.1 as well as from the sarcoplasmic reticulum through the associated ryanodine receptor RyR1. This causes contraction (tensing) of the muscle. Depolarisation of the motor end plate causes potassium ions to leave the muscle cells, repolarising the muscle and closing the calcium channels. Calcium is pumped away from the contractile apparatus and the muscle relaxes.
Mutations altering the usual structure and function of these channels therefore disrupts regulation of muscle contraction, leading to episodes of severe muscle weakness or paralysis. Mutations have been identified in arginine residues making up the voltage sensor of Nav1.4. This voltage sensor comprises the S4 alpha helix of each of the four transmembrane domains (I-IV) of the protein, and contains basic residues that only allow entry of the positive sodium ions at appropriate membrane voltages by blocking or opening the channel pore. In Cav1.1, mutations have also been found in domains II and IV. These mutations are loss-of-function, such that the channels cannot open normally.
In people with mutations in SCN4A or CACNA1S, therefore, the channel has a reduced excitability and signals from the central nervous system are unable to depolarise the muscle. As a result, the muscle cannot contract efficiently (paralysis). The condition is hypokalemic (manifests when potassium is low; not "causing hypokalemia") because a low extracellular potassium ion concentration will cause the muscle to repolarise to the resting potential more quickly, so even if calcium conductance does occur it cannot be sustained. It becomes more difficult to reach the calcium threshold at which the muscle can contract, and even if this is reached then the muscle is more likely to relax. Because of this, the severity would be reduced if potassium ion concentrations are kept high.[7][8][9]
Mutations in KCNJ2 lead to hypokalemic periodic paralysis with cardiac arrhythmias called Andersen–Tawil syndrome.
In contrast, hyperkalemic periodic paralysis refers to gain-of-function mutations in sodium channels that maintain muscle depolarisation and therefore are aggravated by high potassium ion concentrations.
This condition is inherited in an autosomal dominant pattern (but with a high proportion of sporadic cases), which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Diagnosis
Diagnosis can be achieved through a specialized form of electromyographic (EMG) testing called the long exercise test. This test measures the amplitude of a nerve response (called the Compound Muscle Action Potential or CMAP) for 40 to 50 minutes following a few minutes of exercise. In affected people, there is a progressive fall in the amplitude of the potential. Besides the history or a report of serum potassium low normal or low during an attack, the long exercise test is the current standard for medical testing. Genetic diagnosis is often unreliable as only a few of the more common gene locations are tested, but even with more extensive testing 20–37% of people with a clinical diagnosis of hypokalemic periodic paralysis have no known mutation in the two known genes.[10] Standard EMG testing cannot diagnose a person unless they are in a full blown attack at the time of testing. Provoking an attack with exercise and diet then trying oral potassium can be diagnostic, but also dangerous as this form of PP has an alternate form known as hyperkalemic periodic paralysis. The symptoms are almost the same, but the treatment is different. The old glucose insulin challenge is dangerous and risky to the point of being life-threatening and should never be done when other options are so readily available. Factors known to trigger episodes are: stress, cold environment or hypothermia, carbohydrate load, infection, glucose infusion, metabolic alkalosis, alcohol, strenuous exercise, and steroids.
People with hypokalemic periodic paralysis are often misdiagnosed as having a conversion disorder or hysterical paralysis since the weakness is muscle-based and doesn't correspond to nerve or spinal root distributions. The tendency of people with hypokalemic periodic paralysis to get paralyzed when epinephrine is released in "fight or flight" situations further adds to the temptation to misdiagnose the disorder as psychiatric.[11]
Treatment
Treatment of hypokalemic periodic paralysis focuses on preventing further attacks and relieving acute symptoms. Avoiding carbohydrate-rich meals, strenuous exercise and other identified triggers, and taking acetazolamide or another carbonic anhydrase inhibitor, may help prevent attacks of weakness. Some people also take potassium-sparing diuretics such as spironolactone to help maintain potassium levels.[12]
Paralysis attacks can be managed by drinking one of various potassium salts dissolved in water (debate exists over which, if any one in particular, is best used, but potassium chloride and bicarbonate are common). Rapidly absorbed boluses of liquid potassium are generally needed to abort an attack, but some people also find positive maintenance results with time-released potassium tablets. IV potassium is seldom justified unless the person is unable to swallow. Daily potassium dosage may need to be much higher than for potassium replacement from simple hypokalemia: 100-150 mEqs of potassium is often needed to manage daily fluctuations in muscle strength and function.
Perioperatively, prevention includes avoiding neuromuscular blockade, avoid excessive hyperventilation, warm the person, provide adequate hydration, avoid glucose infusions, do not give diuretics, and closely monitor the electrocardiogram for signs of hypokalemia. Normal saline is the preferred IV solution for people with familial hypokalemic periodic paralysis. Glucose containing solutions may cause weakness. Additionally, the high chloride content can cause a mild acidosis which would be preferred over alkalosis.
Prognosis
The prognosis for periodic paralysis varies. Overactivity, a diet that is not low in sodium and carbohydrates, or simply an unfortunate gene mutation can lead to a type of chronic, low level weakness called an "abortive attack," or to permanent muscle damage. Abortive attacks often respond to extra potassium, cutting carbohydrates, getting plenty of rest, increasing doses of medication and gentle daily exercise such as short walks. Permanent muscle weakness is just what it sounds like: Permanent, irreparable damage to the muscles and associated weakness. Vacuoles and tubular aggregates form in and destroy healthy muscle tissue. This type of damage can typically be observed via a muscle biopsy. Not even anabolic steroids can repair this type of muscular damage.
Life span is typically normal normal,[13] but attacks can drop potassium to levels low enough to cause life-threatening breathing problems or heart arrhythmia. People often report muscle pain and cognitive problems during attacks. Migraines occur in up to 50% of all people with hypokalemic periodic paralysiss and may include less common symptoms like phantom smells, sensitivity to light and sound or loss of words. Medical literatures states that muscle strength is normal between attacks, but people often report that their baseline strength is in fact lower than that of healthy individuals.
Because there are dozens of possible gene mutations, some drugs and treatments that work fine for one person will not work for another. For example, most people do well on acetazolamide, but some don't. Some people will do well with extra magnesium, while these same nutrients will make other worse. Care should be take with all new drugs and treatment plans.
History
In 1935 the Scottish physician Dr Mary Walker was the first to recognise the association between familial periodical paralysis and hypokalaemia. She also described the glucose challenge test used in diagnosing hypokalaemic periodic paralysis and the use of intravenous potassium in its treatment.[14][15][16]
See also
References
- ↑ Harrison's principles of internal medicine. Jameson, J. Larry; Kasper, Dennis L.; Longo, Dan L. (Dan Louis), 1949-; Fauci, Anthony S., 1940-; Hauser, Stephen L.; Loscalzo, Joseph (20th ed.). New York. 13 August 2018. p. 307. ISBN 978-1-259-64403-0. OCLC 1029074059.
{{cite book}}: CS1 maint: others (link) - 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 Phuyal, P; Nagalli, S (January 2020). "Hypokalemic Periodic Paralysis". PMID 32644604.
{{cite journal}}: Cite journal requires|journal=(help) - 1 2 Weber, F; Lehmann-Horn, F; Adam, MP; Ardinger, HH; Pagon, RA; Wallace, SE; Bean, LJH; Stephens, K; Amemiya, A (2018). "Hypokalemic Periodic Paralysis". GeneReviews. PMID 20301512.
- ↑ "Hypokalemic periodic paralysis: MedlinePlus Genetics". medlineplus.gov. U.S. National Library of Medicine. Archived from the original on 27 October 2020. Retrieved 26 October 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ↑ Jurkat-Rott, K.; Lehmann-Horn, F. (April 2007). "Genotype-phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis". Neurotherapeutics. 4 (2): 216–24. doi:10.1016/j.nurt.2007.02.001. PMID 17395131.
- ↑ Online Mendelian Inheritance in Man (OMIM): 604433
- ↑ Rüdel R, Lehmann-Horn F, Ricker K, Küther G (February 1984). "Hypokalemic periodic paralysis: in vitro investigation of muscle fiber membrane parameters". Muscle Nerve. 7 (2): 110–20. doi:10.1002/mus.880070205. PMID 6325904. S2CID 25705002.
- ↑ Jurkat-Rott K, Lehmann-Horn F (August 2005). "Muscle channelopathies and critical points in functional and genetic studies". J. Clin. Invest. 115 (8): 2000–9. doi:10.1172/JCI25525. PMC 1180551. PMID 16075040.
- ↑ Kim, SJ; Lee, YJ; Kim, JB (January 2010). "Reduced expression and abnormal localization of the KATP channel subunit SUR2A in patients with familial hypokalemic periodic paralysis". Biochemical and Biophysical Research Communications. 391 (1): 974–8. doi:10.1016/j.bbrc.2009.11.177. PMID 19962959.
- ↑ ""Sternberg D et al. (2009) Hypokalemic Periodic Paralysis, in GeneReviews "". Archived from the original on 2017-01-18. Retrieved 2017-08-31.
- ↑ ""Segal MM, Jurkat-Rott K, Levitt J, Lehmann-Horn F, Hypokalemic periodic paralysis - an owner's manual"". Archived from the original on 2013-12-03. Retrieved 2011-11-24.
- ↑ Kim, JB; Kim, MH (December 2007). "The Genotype and Clinical Phenotype of Korean Patients with Familial Hypokalemic Periodic Paralysis". J Korean Med Sci. 22 (6): 946–51. doi:10.3346/jkms.2007.22.6.946. PMC 2694642. PMID 18162704.
- ↑ Finsterer, J. (2008-03-01). "Primary periodic paralyses". Acta Neurologica Scandinavica. 117 (3): 145–158. doi:10.1111/j.1600-0404.2007.00963.x. ISSN 1600-0404. PMID 18031562. S2CID 22496999.
- ↑ Walker MB (1935). "Potassium chloride in myasthenia gravis". Lancet. 2 (5836): 47. doi:10.1016/S0140-6736(01)09382-5.
- ↑ Encyclopedia of the Neurological Sciences. Academic Press. 2014-04-29. ISBN 9780123851581. Archived from the original on 2021-08-28. Retrieved 2020-11-12.
- ↑ Aitken RS, Allot EN, Gastelden LI, Walker MB (1937). "Observations on a case of familial periodic paralysis". Clin Sci. 3: 47–57.
External links
- GeneReview/NIH/UW entry on Hypokalemic Periodic Paralysis Archived 2017-01-18 at the Wayback Machine
- Levitt JO (2008). "Practical aspects in the management of hypokalemic periodic paralysis". J Transl Med. 6: 18. doi:10.1186/1479-5876-6-18. PMC 2374768. PMID 18426576.
| Classification | |
|---|---|
| External resources |
|