Familial hemiplegic migraine
| Familial hemiplegic migraine | |
|---|---|
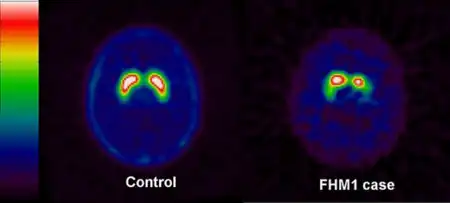 | |
| Dopamine transporter (DAT) imaging with single-photon emission computed tomography -images of a healthy control left and in the FHM1 case right | |
Familial hemiplegic migraine (FHM) is an autosomal dominant type of hemiplegic migraine that typically includes weakness of half the body which can last for hours, days, or weeks. It can be accompanied by other symptoms, such as ataxia, coma, and paralysis. Migraine attacks may be provoked by minor head trauma. Some cases of minor head trauma in patients with hemiplegic migraine can develop into delayed cerebral edema, a life-threatening medical emergency.[1] Clinical overlap occurs in some FHM patients with episodic ataxia type 2 and spinocerebellar ataxia type 6, benign familial infantile epilepsy, and alternating hemiplegia of childhood.
Three genetic loci for FHM are known. FHM1, which accounts for about 50% of FHM patients, is caused by mutations in a gene coding for the P/Q-type calcium channel α subunit, CACNA1A. FHM1 is also associated with cerebellar degeneration. FHM2, which accounts for less than 25% of cases, is caused by mutations in the Na+
/K+
-ATPase gene ATP1A2. FHM3 is a rare subtype of FHM and is caused by mutations in a sodium channel α-subunit coding gene, SCN1A. These three subtypes do not account for all cases of FHM, suggesting the existence of at least one other locus (FHM4).
Also, nonfamilial cases of hemiplegic migraine are seen, termed sporadic hemiplegic migraine. These cases seem to have the same causes as the familial cases and represent de novo mutations. Sporadic cases are also clinically identical to familial cases with the exception of a lack of known family history of attacks.
Signs and symptoms
FHM signs overlap significantly with those of migraine with aura. In short, FHM is typified by migraine with aura associated with hemiparesis, and in FHM1, cerebellar degeneration, which can result in episodic or progressive ataxia. FHM can also present with the same signs as benign familial infantile convulsions and alternating hemiplegia of childhood. Other symptoms are altered consciousness (in fact, some cases seem related to head trauma), gaze-evoked nystagmus, and coma. Aura symptoms, such as numbness and blurring of vision, typically persist for 30–60 minutes, but can last for weeks to months. An attack resembles a stroke, but unlike a stroke, it resolves in time. These signs typically first manifest themselves in the first or second decade of life.
Causes
See the equivalent section in the main migraine article.
FHM mutations are believed to lead to migraine susceptibility by lowering the threshold for cortical-spreading-depression generation. The FHM1 and FHM3 mutations occur in ion channels expressed in neurons. These mutations may lead to both the hyper- and hypoexcitable neurons that might underlie cortical-spreading-depression. How the mutations seen in FHM2 patients might lead to FHM symptoms is even less clear, as the gene mutated in FHM2 is expressed primarily in astrocytes. One proposal states that the depolarization of astrocytes caused by haploinsufficiency of the ATP1A2 Na+
/K+
-ATPase causes increased release of compounds such as adenosine from astrocytes. These compounds then interact with neighboring neurons, altering their excitability and leading to cortical-spreading-depression and migraine.
Pathophysiology
FHM1 (CACNA1A)
The first discovered FHM locus was the CACNA1A gene (originally named CACNL1A4), which encodes the P/Q-type calcium channel CaV2.1. Currently, 17 mutations in this channel are known (table 1), and these mutations are distributed throughout the channel. Some of these mutations result in patients with notable cerebellar degeneration or other dysfunction, including one mutation (S218L), which may be related to severe responses to mild concussion, up to and including delayed cerebral edema, coma, and death.[2] Fifteen of these mutants have received at least some further analysis at the electrophysiological level to attempt to determine how they might lead to the FHM1 phenotype. Contradiction in the literature is increasing as to the end result of these mutations on channel kinetics and neuronal excitability.
A good example of this contradiction can be seen in the literature regarding the R192Q mutation.[3] The first investigation of this mutation, using the rabbit isoform of the channel expressed in oocytes, found that it did not alter any measured channel properties.[4] A subsequent report, using human channels expressed in HEK293 cells, found a small, hyperpolarizing shift in the midpoint for activation, a result common among FHM1 mutants.[5] This shift results in channels that open at more negative potentials, thus have a higher open probability than wild-type channels at most potentials. This report also found that the R192Q mutant produced almost twice as much whole-cell current compared to wild-type channels. This is not due to a change in single channel conductance, but to an equivalent increase in channel density. A subsequent group noticed that this mutation is in a region important for modulation by G protein-coupled receptors (GPCRs).[6] GPCR activation leads to inhibition of wild-type CaV2.1 currents. R192Q mutant channel currents are also decreased by GPCR activation, but by a smaller amount. A more recent group has confirmed some of these results by creating a R192Q knock-in mouse.[7] They confirmed that the R192Q mutant activates at more negative potentials and that neurons producing these channels have much larger whole-cell current. This resulted in a much larger quantal content (the number of neurotransmitter packets released per action potential) and generally enhanced neurotransmitter release in R192Q-expressing neurons versus wild-type. Consequently, these mutant mice were more susceptible to cortical-spreading-depression than their wild-type counterparts. The most recent experiments on this mutant, however, have contradicted some of these results.[8] In CaV2.1 knockout neurons transfected with human channels, P/Q-type currents from mutant channels are actually smaller than their wild-type counterpart. They also found a significant decrease in calcium influx during depolarization, leading to decreased quantal content, in mutant versus wild-type expressing neurons. Neurons expressing mutant channels were also less able to mediate inhibitory input and have smaller inhibitory postsynaptic currents through P/Q-type channels. Further testing with this and other mutants is required to determine their end effect on human physiology.
| Mutation | Position | Effect | Cerebellar signs | Reference | |
|---|---|---|---|---|---|
| Nucleotide | Amino acid | ||||
| c.G575A | R192Q | D1S4 | Decreases G-protein mediated inhibition, activates at more negative potentials, increased expression, faster recovery from inactivation. In mice: greater current, activates at more negative potentials, enhances transmitter release | ? | [3][4][5][6][7][8] |
| c.G584A | R195K | D1S4 | No | [9] | |
| c.C653T | S218L | D1S4-5 | Increases sojourns to subconductances, activates at more negative potentials, decreased slow inactivation, increased fast inactivation | Yes | [2][10] |
| c.G1748A | R583Q* | D2S4 | Activates at more negative potentials, faster current decay, faster inactivation, slower recovery from inactivation | Yes | [9][11][12][13][14] |
| c.C1997T | T666M | D2-pore | Activates at more negative potentials, faster current decay, slowed recovery from inactivation, smaller single channel conductance, higher i*Po, slower recovery from inactivation, Increased G-protein mediated inhibition, decreased gating charge (fewer channels available to open) | Yes | [3][4][5][8][9][13][15][16][17] |
| c.T2141C | V714A | D2S6 | Activates at more negative potentials, faster current decay, faster recovery from inactivation, decreases expression, faster recovery from inactivation, increases G-protein-mediated inhibition | No | [3][4][5][8][13] |
| c.C2145G | D715E | D2S6 | Activates at more negative potentials, faster current decay, faster inactivation | Yes | [9][11][15] |
| c.A4003G | K1335E | D3S3-4 | Activates at more negative potentials, inactivates at more negative potentials, slowed recovery from inactivation, increased frequency dependent rundown | No | [9][18] |
| c.G4037A | R1346Q | D3S4 | Yes | [19] | |
| c.A4151G | Y1384C | D3S5 | Yes | [9][20] | |
| c.G4366T | V1456L | D3-pore | Activates at more negative potentials, slower current decay, slower recovery from inactivation | No | [12][21] |
| c.C4636T | R1546X** | D4S1 | Decreased current | Yes | [22][23][24] |
| c.C4999T | R1667W | D4S4 | Yes | [9] | |
| c.T5047C | W1683R | D4S4-5 | Activates at more negative potentials, inactivates at more negative potentials, slowed recovery from inactivation, increased frequency dependent rundown | Yes | [9][18] |
| c.G5083A | V1695I | D4S5 | Slowed recovery from inactivation, increased frequency dependent rundown | No | [9][18] |
| c.T5126C | I1709T | D4S5 | Yes | [25][26] | |
| c.A5428C | I1810L | D4S6 | Activates at more negative potentials, faster recovery from inactivation, decreased expression, faster recovery from inactivation, Increased G-protein mediated inhibition | Yes | [3][4][5][8][13] |
* ** |
Also diagnosed as spinocerebellar ataxia type-6 Also diagnosed as episodic ataxia type-2 | ||||
| Sequence numbering according to NCBI reference sequence NM_000068.2 Archived 2022-09-20 at the Wayback Machine. Cerebellar signs refers to findings of cerebellar degeneration or ataxia upon clinical examination. | |||||
FHM2 (ATP1A2)
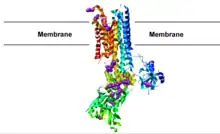
/K+
-ATPase with FHM2 mutations noted in purple: The N-terminus is colored blue and the C-terminus red. The approximate location of the cell membrane is noted. The original pdb file is available here.
The second subtype of familial hemiplegic migraine, FHM2, is caused by mutations in the gene ATP1A2 that encodes a Na+
/K+
-ATPase. This Na+
/K+
-ATPase is heavily expressed in astrocytes and helps to set and maintain their reversal potential. Twenty-nine known mutations in this gene are associated with FHM2 (table 2), many clustering in the large intracellular loop between membrane-spanning segments 4 and 5 (figure 1). Twelve of these mutations have been studied by expression in model cells. All but one have shown either complete loss of function or more complex decreases in ATPase activity or potassium sensitivity. Astrocytes expressing these mutant ion pumps will have much higher resting potentials and are believed to lead to disease through a poorly understood mechanism.
| Mutation | Location | Physiological result | Reference(s) |
|---|---|---|---|
| E174K | M2-3 | No change | [27] |
| T263M | M2-3 | [28] | |
| G301R | M3 | [29] | |
| T345A | M4-5 | Decreased K influx | [30][31] |
| T376M | M4-5 | [28] | |
| R383H | M4-5 | [32] | |
| T378N | M4-5 | [33] | |
| C515Y | M4-5 | Loss of function (haploinsufficiency) | [27] |
| R548H | M4-5 | [34] | |
| R593W | M4-5 | Loss of function (haploinsufficiency) | [35] |
| A606T | M4-5 | [28] | |
| G615R | M4-5 | Loss of function (haploinsufficiency) | [36] |
| V628M | M4-5 | Loss of function (haploinsufficiency) | [35] |
| R689Q | M4-5 | Decreased catalytic turnover | [31][37][38] |
| E700K | M4-5 | [39] | |
| D718N | M4-5 | Loss of function (haploinsufficiency) | [32] |
| M731T | M4-5 | Decreased catalytic turnover | [31][37][38] |
| R763H | M4-5 | Loss of function (haploinsufficiency) | [32] |
| L764P | M4-5 | Loss of function (haploinsufficiency) | [31][40][41] |
| P796R | M5-6 | [32] | |
| M829R | M6-7 | [28] | |
| R834Q | M6-7 | [28] | |
| W887R | M7-8 | Loss of function (haploinsufficiency) | [27][31][40][41] |
| E902K | M7-8 | [32] | |
| 935K_940SdelinsI | M8-9 | [28] | |
| R937P | M8-9 | [28] | |
| S966LfsX998 | M9 | [28] | |
| P979L | M9-10 | [32] | |
| X1021RextX28 | C-Terminus | [32] | |
| Numbering according to the NCBI reference sequence NM_000702.2 Archived 2022-09-20 at the Wayback Machine. | |||
FHM3 (SCN1A)
The final known locus FHM3 is the SCN1A gene, which encodes a sodium channel α subunit. The only study so far that has found mutations in this gene discovered the same Q1489K mutation in three of 20 families (15%) with 11 other kindreds (55%) already having mutations in CACNA1A or ATP1A2. This mutation is located in a highly conserved region of an intracellular loop connecting domains three and four. This mutation results in a greatly hastened (two- to four-fold) recovery from inactivation compared to wild-type.[42] As this channel is important for action potential generation in neurons, the Q1489K mutant is expected to result in hyperexcitable neurons.
FHM4 (1q31)
The final locus for FHM maps to the q-arm of chromosome 1. A number of attractive candidate genes occur in this area, though no mutations in them have yet been linked to FHM4.[43]
Other genetic associations
A fourth gene associated with this condition is the proline-rich transmembrane protein 2 (PRRT2 gene) - an axonal protein associated with the exocytosis complex.[44]
A fifth gene associated with this condition is SLC4A4, which encodes the electrogenic NaHCO3 cotransporter NBCe1.[45]
Diagnosis
Diagnosis of FHM is made according to these criteria:
- Two attacks of each of:
- Aura with motor weakness accompanied by either reversible visual symptoms (flickering lights, spots, lines, etc.), reversible sensory symptoms (pins and needles, numbness, etc.) or speech symptoms
- At least two occurrences of:
- One or more aura symptoms that develop over at least 5 minutes
- These symptoms lasting more than 5 minutes and less than 24 hours
- Headache beginning within 60 minutes of aura onset: These headaches can last 4–72 hours, occur on only one side of the head, pulsate, be of moderate to severe intensity, and may be aggravated by common physical activities such as walking. These headaches must also be accompanied by nausea/vomiting, phonophobia (avoidance of sound due to hypersensitivity), and/or photophobia (avoidance of light due to hypersensitivity).
- At least one close (first- or second-degree) relative with FHM
- No other likely cause
Sporadic forms follow the same diagnostic criteria, with the exception of family history.
In all cases, family and patient histories are used for diagnosis. Brain-imaging techniques, such as MRI, CAT scan, and SPECT,[46] are used to look for signs of other familial conditions such as CADASIL or mitochondrial disease, and for evidence of cerebellar degeneration. With the discovery of causative genes, genetic sequencing can also be used to verify diagnosis (though not all genetic loci are known).
Screening
Prenatal screening is not typically done for FHM, but it may be performed if requested. As penetrance is high, individuals found to carry mutations should be expected to develop signs of FHM at some point in life.
Management
See the equivalent section in the main migraine article.
People with FHM are encouraged to avoid activities that may trigger their attacks. Minor head trauma is a common attack precipitant, so FHM sufferers should avoid contact sports. Acetazolamide or standard drugs are often used to treat attacks, though those leading to vasoconstriction should be avoided due to the risk of stroke.
Epidemiology
Migraine itself is a very common disorder, occurring in 15–20% of the population. Hemiplegic migraine, be it familial or spontaneous, is less prevalent, at 0.01% prevalence according to one report.[47] Women are three times more likely to be affected than males.
See also
References
- ↑ Stam AH, Luijckx GJ, Poll-Thé BT, Ginjaar IB, Frants RR, Haan J, Ferrari MD, Terwindt GM, van den Maagdenberg AM (2009). "Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation". J Neurol Neurosurg Psychiatry. 80 (10): 1125–1129. doi:10.1136/jnnp.2009.177279. PMID 19520699. S2CID 1000464. Archived from the original on 2022-09-20. Retrieved 2022-04-08.
- 1 2 Kors E, Terwindt G, Vermeulen F, Fitzsimons R, Jardine P, Heywood P, Love S, van den Maagdenberg A, Haan J, Frants R, Ferrari M (2001). "Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine". Ann Neurol. 49 (6): 753–60. doi:10.1002/ana.1031. PMID 11409427. S2CID 23340019.
- 1 2 3 4 5 Ophoff R, Terwindt G, Vergouwe M, van Eijk R, Oefner P, Hoffman S, Lamerdin J, Mohrenweiser H, Bulman D, Ferrari M, Haan J, Lindhout D, van Ommen G, Hofker M, Ferrari M, Frants R (1996). "Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+
channel gene CACNL1A4". Cell. 87 (3): 543–52. doi:10.1016/S0092-8674(00)81373-2. hdl:1765/57576. PMID 8898206. S2CID 16840573. Archived from the original on 2022-03-27. Retrieved 2022-04-08. - 1 2 3 4 5 Kraus R, Sinnegger M, Glossmann H, Hering S, Striessnig J (1998). "Familial hemiplegic migraine mutations change alpha1A Ca2+
channel kinetics". J Biol Chem. 273 (10): 5586–90. doi:10.1074/jbc.273.10.5586. PMID 9488686. - 1 2 3 4 5 Hans M, Luvisetto S, Williams M, Spagnolo M, Urrutia A, Tottene A, Brust P, Johnson E, Harpold M, Stauderman K, Pietrobon D (1999). "Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine". J Neurosci. 19 (5): 1610–9. doi:10.1523/JNEUROSCI.19-05-01610.1999. PMC 6782159. PMID 10024348.
- 1 2 Melliti K, Grabner M, Seabrook G (2003). "The familial hemiplegic migraine mutation R192q reduces G-protein-mediated inhibition of p/q-type (Cav2.1) calcium channels expressed in human embryonic kidney cells". J Physiol. 546 (Pt 2): 337–47. doi:10.1113/jphysiol.2002.026716. PMC 2342512. PMID 12527722.
- 1 2 van den Maagdenberg A, Pietrobon D, Pizzorusso T, Kaja S, Broos L, Cesetti T, van de Ven R, Tottene A, van der Kaa J, Plomp J, Frants R, Ferrari M (2004). "A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression". Neuron. 41 (5): 701–10. doi:10.1016/S0896-6273(04)00085-6. PMID 15003170. S2CID 1456789.
- 1 2 3 4 5 Cao Y, Tsien R (2005). "Effects of familial hemiplegic migraine type 1 mutations on neuronal P/Q-type Ca2+
channel activity and inhibitory synaptic transmission". Proc Natl Acad Sci USA. 102 (7): 2590–5. doi:10.1073/pnas.0409896102. PMC 548328. PMID 15699344. - 1 2 3 4 5 6 7 8 9 Ducros A, Denier C, Joutel A, Cecillon M, Lescoat C, Vahedi K, Darcel F, Vicaut E, Bousser M, Tournier-Lasserve E (2001). "The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel". N Engl J Med. 345 (1): 17–24. doi:10.1056/NEJM200107053450103. PMID 11439943.
- ↑ Tottene A, Pivotto F, Fellin T, Cesetti T, van den Maagdenberg A, Pietrobon D (2005). "Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma". J Biol Chem. 280 (18): 17678–86. doi:10.1074/jbc.M501110200. PMID 15743764.
- 1 2 Battistini S, Stenirri S, Piatti M, Gelfi C, Righetti P, Rocchi R, Giannini F, Battistini N, Guazzi G, Ferrari M, Carrera P (1999). "A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia". Neurology. 53 (1): 38–43. doi:10.1212/WNL.53.1.38. PMID 10408534. S2CID 32390280.
- 1 2 Kraus R, Sinnegger M, Koschak A, Glossmann H, Stenirri S, Carrera P, Striessnig J (2000). "Three new familial hemiplegic migraine mutants affect P/Q-type Ca(2+) channel kinetics". J Biol Chem. 275 (13): 9239–43. doi:10.1074/jbc.275.13.9239. PMID 10734061.
- 1 2 3 4 Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, Pietrobon D (2002). "Familial hemiplegic migraine mutations increase Ca2+
influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons". Proc Natl Acad Sci USA. 99 (20): 13284–9. doi:10.1073/pnas.192242399. PMC 130625. PMID 12235360. - ↑ Alonso I, Barros J, Tuna A, Coelho J, Sequeiros J, Silveira I, Coutinho P (2003). "Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic migraine caused by a unique CACNA1A missense mutation in patients from a large family". Arch Neurol. 60 (4): 610–4. doi:10.1001/archneur.60.4.610. PMID 12707077.
- 1 2 Ducros A, Denier C, Joutel A, Vahedi K, Michel A, Darcel F, Madigand M, Guerouaou D, Tison F, Julien J, Hirsch E, Chedru F, Bisgård C, Lucotte G, Després P, Billard C, Barthez M, Ponsot G, Bousser M, Tournier-Lasserve E (1999). "Recurrence of the T666M calcium channel CACNA1A gene mutation in familial hemiplegic migraine with progressive cerebellar ataxia". Am J Hum Genet. 64 (1): 89–98. doi:10.1086/302192. PMC 1377706. PMID 9915947.
- ↑ Friend K, Crimmins D, Phan T, Sue C, Colley A, Fung V, Morris J, Sutherland G, Richards R (1999). "Detection of a novel missense mutation and second recurrent mutation in the CACNA1A gene in individuals with EA-2 and FHM". Hum Genet. 105 (3): 261–5. doi:10.1007/s004390051099. PMID 10987655.
- ↑ Barrett C, Cao Y, Tsien R (2005). "Gating deficiency in a familial hemiplegic migraine type 1 mutant P/Q-type calcium channel". J Biol Chem. 280 (25): 24064–71. doi:10.1074/jbc.M502223200. PMID 15795222.
- 1 2 3 Müllner C, Broos L, van den Maagdenberg A, Striessnig J (2004). "Familial hemiplegic migraine type 1 mutations K1336E, W1684R, and V1696I alter Cav2.1 Ca2+
channel gating: evidence for beta-subunit isoform-specific effects". J Biol Chem. 279 (50): 51844–50. doi:10.1074/jbc.M408756200. PMID 15448138. - ↑ Alonso I, Barros J, Tuna A, Seixas A, Coutinho P, Sequeiros J, Silveira I (2004). "A novel R1347Q mutation in the predicted voltage sensor segment of the P/Q-type calcium-channel alpha-subunit in a family with progressive cerebellar ataxia and hemiplegic migraine". Clin Genet. 65 (1): 70–2. doi:10.1111/j..2004.00187.x. PMID 15032980. S2CID 45747611.
- ↑ Vahedi K, Denier C, Ducros A, Bousson V, Levy C, Chabriat H, Haguenau M, Tournier-Lasserve E, Bousser M (2000). "CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy". Neurology. 55 (7): 1040–2. doi:10.1212/WNL.55.7.1040. PMID 11061267. S2CID 26855561.
- ↑ Carrera P, Piatti M, Stenirri S, Grimaldi L, Marchioni E, Curcio M, Righetti P, Ferrari M, Gelfi C (1999). "Genetic heterogeneity in Italian families with familial hemiplegic migraine". Neurology. 53 (1): 26–33. doi:10.1212/WNL.53.1.26. PMID 10408532. S2CID 25604212.
- ↑ Denier C, Ducros A, Vahedi K, Joutel A, Thierry P, Ritz A, Castelnovo G, Deonna T, Gérard P, Devoize J, Gayou A, Perrouty B, Soisson T, Autret A, Warter J, Vighetto A, Van Bogaert P, Alamowitch S, Roullet E, Tournier-Lasserve E (1999). "High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2". Neurology. 52 (9): 1816–21. doi:10.1212/WNL.52.9.1816. PMID 10371528. S2CID 39421883.
- ↑ Jen J, Yue Q, Nelson S, Yu H, Litt M, Nutt J, Baloh R (1999). "A novel nonsense mutation in CACNA1A causes episodic ataxia and hemiplegia". Neurology. 53 (1): 34–7. doi:10.1212/WNL.53.1.34. PMID 10408533. S2CID 22046224.
- ↑ Jen J, Wan J, Graves M, Yu H, Mock A, Coulin C, Kim G, Yue Q, Papazian D, Baloh R (2001). "Loss-of-function EA2 mutations are associated with impaired neuromuscular transmission". Neurology. 57 (10): 1843–8. doi:10.1212/WNL.57.10.1843. PMID 11723274. S2CID 23258478.
- ↑ Beauvais K, Cavé-Riant F, De Barace C, Tardieu M, Tournier-Lasserve E, Furby A (2004). "New CACNA1A gene mutation in a case of familial hemiplegic migraine with status epilepticus". Eur Neurol. 52 (1): 58–61. doi:10.1159/000079546. PMID 15240985. S2CID 35019951.
- ↑ Kors E, Vanmolkot K, Haan J, Kheradmand Kia S, Stroink H, Laan L, Gill D, Pascual J, van den Maagdenberg A, Frants R, Ferrari M (2004). "Alternating hemiplegia of childhood: no mutations in the second familial hemiplegic migraine gene ATP1A2". Neuropediatrics. 35 (5): 293–6. doi:10.1055/s-2004-821082. PMID 15534763.
- 1 2 3 Todt U, Dichgans M, Jurkat-Rott K, Heinze A, Zifarelli G, Koenderink J, Goebel I, Zumbroich V, Stiller A, Ramirez A, Friedrich T, Göbel H, Kubisch C (2005). "Rare missense variants in ATP1A2 in families with clustering of common forms of migraine". Hum Mutat. 26 (4): 315–21. doi:10.1002/humu.20229. PMID 16110494. S2CID 15851518.
- 1 2 3 4 5 6 7 8 Riant F, De Fusco M, Aridon P, Ducros A, Ploton C, Marchelli F, Maciazek J, Bousser M, Casari G, Tournier-Lasserve E (2005). "ATP1A2 mutations in 11 families with familial hemiplegic migraine". Hum Mutat. 26 (3): 281. doi:10.1002/humu.9361. PMID 16088919.
- ↑ Spadaro M, Ursu S, Lehmann-Horn F, Veneziano L, Liana V, Antonini G, Giovanni A, Giunti P, Paola G, Frontali M, Jurkat-Rott K (2004). "A G301R Na+
/K+
-ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs". Neurogenetics. 5 (3): 177–85. doi:10.1007/s10048-004-0183-2. PMID 15459825. S2CID 1480291. - ↑ Kaunisto M, Harno H, Vanmolkot K, Gargus J, Sun G, Hämäläinen E, Liukkonen E, Kallela M, van den Maagdenberg A, Frants R, Färkkilä M, Palotie A, Wessman M (2004). "A novel missense ATP1A2 mutation in a Finnish family with familial hemiplegic migraine type 2". Neurogenetics. 5 (2): 141–6. doi:10.1007/s10048-004-0178-z. PMID 15133718. S2CID 27379120.
- 1 2 3 4 5 Segall L, Scanzano R, Kaunisto M, Wessman M, Palotie A, Gargus J, Blostein R (2004). "Kinetic alterations due to a missense mutation in the Na,K-ATPase alpha2 subunit cause familial hemiplegic migraine type 2". J Biol Chem. 279 (42): 43692–6. doi:10.1074/jbc.M407471200. PMID 15308625.
- 1 2 3 4 5 6 7 Jurkat-Rott K, Freilinger T, Dreier J, Herzog J, Göbel H, Petzold G, Montagna P, Gasser T, Lehmann-Horn F, Dichgans M (2004). "Variability of familial hemiplegic migraine with novel A1A2 Na+
/K+
-ATPase variants". Neurology. 62 (10): 1857–61. doi:10.1212/01.WNL.0000127310.11526.FD. PMID 15159495. S2CID 43023377. - ↑ Swoboda K, Kanavakis E, Xaidara A, Johnson J, Leppert M, Schlesinger-Massart M, Ptacek L, Silver K, Youroukos S (2004). "Alternating hemiplegia of childhood or familial hemiplegic migraine? A novel ATP1A2 mutation". Ann Neurol. 55 (6): 884–7. doi:10.1002/ana.20134. PMID 15174025. S2CID 13430399.
- ↑ Ambrosini A, D'Onofrio M, Grieco G, Di Mambro A, Montagna G, Fortini D, Nicoletti F, Nappi G, Sances G, Schoenen J, Buzzi M, Santorelli F, Pierelli F (2005). "Familial basilar migraine associated with a new mutation in the ATP1A2 gene". Neurology. 65 (11): 1826–8. doi:10.1212/01.wnl.0000187072.71931.c0. PMID 16344534. S2CID 12870819.
- 1 2 Vanmolkot K, Kors E, Turk U, Turkdogan D, Keyser A, Broos L, Kia S, van den Heuvel J, Black D, Haan J, Frants R, Barone V, Ferrari M, Casari G, Koenderink J, van den Maagdenberg A (2006). "Two de novo mutations in the Na,K-ATPase gene ATP1A2 associated with pure familial hemiplegic migraine". Eur J Hum Genet. 14 (5): 555–60. doi:10.1038/sj.ejhg.5201607. PMID 16538223.
- ↑ Vanmolkot K, Stroink H, Koenderink J, Kors E, van den Heuvel J, van den Boogerd E, Stam A, Haan J, De Vries B, Terwindt G, Frants R, Ferrari M, van den Maagdenberg A (2006). "Severe episodic neurological deficits and permanent mental retardation in a child with a novel FHM2 ATP1A2 mutation". Ann Neurol. 59 (2): 310–4. doi:10.1002/ana.20760. PMID 16437583. S2CID 8626672.
- 1 2 Vanmolkot K, Kors E, Hottenga J, Terwindt G, Haan J, Hoefnagels W, Black D, Sandkuijl L, Frants R, Ferrari M, van den Maagdenberg A (2003). "Novel mutations in the Na+
, K+
-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions". Ann Neurol. 54 (3): 360–6. doi:10.1002/ana.10674. PMID 12953268. S2CID 43526424. - 1 2 Segall L, Mezzetti A, Scanzano R, Gargus J, Purisima E, Blostein R (2005). "Alterations in the α2 isoform of Na,K-ATPase associated with familial hemiplegic migraine type 2". Proc Natl Acad Sci USA. 102 (31): 11106–11. Bibcode:2005PNAS..10211106S. doi:10.1073/pnas.0504323102. PMC 1178013. PMID 16037212.
- ↑ Pierelli F, Grieco G, Pauri F, Pirro C, Fiermonte G, Ambrosini A, Costa A, Buzzi M, Valoppi M, Caltagirone C, Nappi G, Santorelli F (2006). "A novel ATP1A2 mutation in a family with FHM type II". Cephalalgia. 26 (3): 324–8. doi:10.1111/j.1468-2982.2006.01002.x. PMID 16472340. S2CID 33708885.
- 1 2 De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, Ballabio A, Aridon P, Casari G (2003). "Haploinsufficiency of ATP1A2 encoding the Na+
/K+
pump alpha2 subunit associated with familial hemiplegic migraine type 2". Nat Genet. 33 (2): 192–6. doi:10.1038/ng1081. PMID 12539047. S2CID 1296597. - 1 2 Koenderink J, Zifarelli G, Qiu L, Schwarz W, De Pont J, Bamberg E, Friedrich T (2005). "Na,K-ATPase mutations in familial hemiplegic migraine lead to functional inactivation". Biochim Biophys Acta. 1669 (1): 61–8. doi:10.1016/j.bbamem.2005.01.003. PMID 15843000.
- ↑ Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari M, Herzog J, van den Maagdenberg A, Pusch M, Strom T (2005). "Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine". Lancet. 366 (9483): 371–7. doi:10.1016/S0140-6736(05)66786-4. PMID 16054936. S2CID 30365311.
- ↑ Gardner K, Barmada M, Ptacek L, Hoffman E (1997). "A new locus for hemiplegic migraine maps to chromosome 1q31". Neurology. 49 (5): 1231–8. doi:10.1212/WNL.49.5.1231. PMID 9371899. S2CID 32611926.
- ↑ Ducros, A (Apr 22, 2013). "[Genetics of migraine.]". Revue Neurologique. 169 (5): 360–71. doi:10.1016/j.neurol.2012.11.010. PMID 23618705.
- ↑ Suzuki, M.; Van Paesschen, W.; Stalmans, I.; Horita, S.; Yamada, H.; Bergmans, B. A.; Legius, E.; Riant, F.; De Jonghe, P.; Li, Y.; Sekine, T.; Igarashi, T.; Fujimoto, I.; Mikoshiba, K.; Shimadzu, M.; Shiohara, M.; Braverman, N.; Al-Gazali, L.; Fujita, T.; Seki, G. (23 August 2010). "Defective membrane expression of the Na+-HCO3- cotransporter NBCe1 is associated with familial migraine". Proceedings of the National Academy of Sciences. 107 (36): 15963–15968. Bibcode:2010PNAS..10715963S. doi:10.1073/pnas.1008705107. PMC 2936614. PMID 20798035.
- ↑ Arias-Rivas S, Rodríguez-Yañez M, Cortés J, Pardo-Parrado M, Aguiar P, Leira R, Castillo J, Blanco M (2012). "Familial hemiplegic migraine with prolonged global aura: follow-up findings of subtraction ictal SPECT co-registered to MRI (SISCOM)". Cephalalgia. 32 (13): 1013–1014. doi:10.1177/0333102412457093. PMID 22933508. S2CID 18838384.
- ↑ Lykke Thomsen L, Kirchmann Eriksen M, Faerch Romer S, Andersen I, Ostergaard E, Keiding N, Olesen J, Russell M (2002). "An epidemiological survey of hemiplegic migraine". Cephalalgia. 22 (5): 361–75. doi:10.1046/j.1468-2982.2002.00371.x. PMID 12110112. S2CID 22040734.
External links
- GeneReviews/NCBI/NIH/UW entry on Familial Hemiplegic Migraine Archived 2022-06-15 at the Wayback Machine
- "The International Classification of Headache Disorders 2nd Edition". Cephalalgia. 24 (s1): 8–160. May 2004. ISSN 1468-2982. Archived from the original on 2022-03-09. Retrieved 2022-04-08.
| Classification | |
|---|---|
| External resources |
|