Carpenter syndrome
| Carpenter syndrome | |
|---|---|
| Other names: Acrocephalopolysyndactyly type II | |
Carpenter syndrome, also called acrocephalopolysyndactyly type II,[1] is an extremely rare autosomal recessive[2] congenital disorder characterized by craniofacial malformations, obesity, and syndactyly.[2]
It was first characterized in 1909, and is named for George Alfred Carpenter.[3][4]
Symptoms and signs

Carpenter syndrome presents several features:[5]
- Tower-shaped skull (craniosynostosis)
- Additional or fused digits (fingers and toes)
- Obesity
- Reduced height
- undescended testes
Intellectual disability is also common with the disorder, although some patients may have average intellectual capacity.[6]
Description
Carpenter Syndrome belongs to a group of rare genetic disorders known as acrocephalopolysyndactyly, abbreviated ACPS (RN, 2007). There were originally five types of ACPS, but this number has been decreased because they have been found to be closely related to one another or to other disorders (Paul A. Johnson, 2002). The most common physical manifestation of Carpenter Syndrome is early fusing of the fibrous cranial sutures which results in an abnormally pointed head. The fusion of the skull bones is evident from birth (National Organization for Rare Disorders, Inc., 2008). Babies’ mobile cranial bones form a cone shape as they pass through the birth canal and soon thereafter return to a normal shape; however, a baby affected by carpenter syndrome maintains a cone shaped head.
A baby affected by Carpenter Syndrome will also display malformations of the face. An individual affected by the syndrome may have broad cheeks, a flat nasal bridge, and a wide upturned nose with abnormally large nasal openings. Their ears will most commonly be low, unevenly set, and malformed in structure. In addition to these facial abnormalities, individuals also have an underdeveloped maxilla and/or mandible with a highly arched and narrow palate which makes speech a very difficult skill to master. Teeth are usually very late to come in and will be undersized and spaced far apart.
Other physical abnormalities often associated with Carpenter Syndrome include extra digits. Extra toes are more commonly seen than fingers. Often both the toes and fingers are webbed, a process that occurs before the sixth week gestational period. Often their digits will be abnormally short, and the fingers are commonly missing an interphalangeal joint. Roughly half of the babies born with Carpenter Syndrome have some type of heart defect, and seventy five percent of individuals with this disease will experience some degree of development delay due to mild mental retardation (Carpenter Syndrome-description).
Genetics
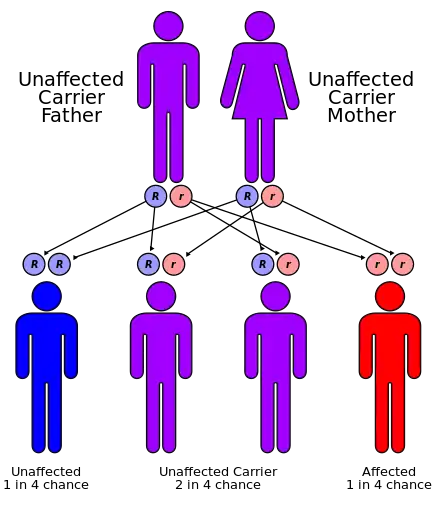
Carpenter syndrome has been associated with mutations in the RAB23 gene,[7] which is located on chromosome 6 in humans. Additionally, three key SNPs in the MEGF8 gene,[8] located on chromosome 19 at 19q13.2, have been identified as primary causes of Carpenter syndrome.
Diagnosis
The diagnosis of Carpenter Syndrome is made based on the presence of the bicoronal and sagittal skull malformations, which results in a pointed, cone-shaped or short, broad head. The diagnosis is also made based on the presence of extra or fused digits. X rays and/ or CT scans of the skull may be performed in order to accurately diagnose the individual; however, other genetic disorders, which have available genetic tests, are also characterized by skull malformations. A positive result on these tests can rule out a Carpenter Syndrome diagnosis.
Types
The primary diagnostic factor is a malformation of the skull. The two most common types of craniosynostosis are sagittal and bicoronal. Sagittal craniosynostosis manifests itself by causing a long narrow skull, resembling a football. It is quantitatively determined by measuring the anterior to posterior (front to back) diameter of the skull. An increased A-P diameter indicates a malformed fusion of the sagittal suture. Individuals affected with sagittal craniosynostosis have narrow, prominent foreheads and the back of the head is much larger than normal. The “soft spot” is very small or missing altogether with this particular type of craniosynostosis.
The second common type of skull malformation is bicoronal craniosynostosis is characterized by a wide, short skull. In this particular type of craniosynostosis the A-P diameter is smaller than in normal individuals. These individuals have malformed eye sockets and foreheads. The eye sockets are much smaller than normal and often cause visual impairment. Complications may include damage to the optic nerve, resulting in a decrease in visual clarity, bulging eyeballs as a result of shallow eye orbits which usually causes some sort of damage to the cornea (the outer layer of the eye). Bicoronal craniosynostosis may also result in widely spaced eyes and narrowing of the sinuses and tear ducts that may result in inflammation of the mucous membranes of the exposed portion of the eye.
In addition to the previously named complications of bicoronal craniosynostosis, many babies will also be affected by hydrocephalus, more commonly known as water on the brain. Hydrocephalus results in increased pressure on the brain which can cause permanent brain damage if not treated promptly. An abnormally highly arched palate is also seen in affected individuals causing dental problems and the thrusting forward of the lower jaw. Individuals affected by Carpenter syndrome often experience cutaneous syndactyly (fusion of the digits) or polydactyly (presence of extra digits) of the toes more often than fingers. Individuals also have short fingers. Approximately one third of individuals born with Carpenter Syndrome have a type of heart defect. Commonly seen heart defects may include: narrowing of the pulmonary artery, transposition of the major blood vessels, or the presence of an abnormally large vena cava, which delivers blood back to the heart from the head, neck, and upper limbs. The testes of males affected by Carpenter Syndrome may also fail to descend (Paul A. Johnson, 2002).
Treatment
Operations to correct the malformations of the skull should be performed within the first year of infancy in patients affected by Carpenter Syndrome. Performing surgery at a young age increases the likelihood of obtaining a greatly improved appearance of the head because modifying bone is much easier to do when the skull is still constantly growing and changing.[9] In surgery the doctor breaks the fused sutures to allow for brain growth. Doctors remove the cranial plates of the skull, reshape them and replace them back onto the skull in an attempt to reshape the head to appear more normal. Although the sutures are broken during surgery they will quickly refuse, and in some cases holes form in the plates allowing cerebral spinal fluid to escape into cyst like structures on the external surface of the head.[10]
If an individual with Carpenter Syndrome has a serious heart defect they will require surgery to correct the malformation of the heart. Other elective surgeries may also be performed. Some parents opt to have their child’s webbed fingers or toes separated which improves their appearance but not necessarily the functionality of the digits. In order to address the occupational challenges of the disease, many children with Carpenter Syndrome go through speech and occupational therapy in order to achieve more independence in everyday tasks and activities (RN, 2007).
In order to address the vision problems that are associated with bicoronal craniosynostosis, the individual must seek consultation from an ophthalmologist. If the palate is severely affected dental consultation may be necessary to correct the malformation. Obesity is often associated with Carpenter Syndrome, so a lifelong diet plan is often utilized to maintain a healthy weight. In addition surgery must be performed if the testes fail to descend (Paul A. Johnson, 2002). If the procedure is not performed the individual will become infertile.
Occurrence
There are approximately three hundred known cases of Carpenter Syndrome in the United States. Only 1 in 1 million live births will result in an infant affected by Carpenter Syndrome (RN, 2007).
Carpenter Syndrome is an autosomal recessive disease which means both parents must have the faulty genes in order to pass the disease onto their children. Even if both parents possess the faulty gene there is still only a twenty five percent chance that they will produce a child affected by the syndrome. Their children who do not have the disease will still be carriers and possess the ability to pass the disease onto their offspring if their spouse is also a carrier of the particular gene.[9]
See also
References
- ↑ Online Mendelian Inheritance in Man (OMIM): 201000
- 1 2 Perlyn, Ca; Marsh, Jl (March 1909). "Craniofacial dysmorphology of Carpenter syndrome: lessons from three affected siblings". Plastic and Reconstructive Surgery. 121 (3): 971–81. doi:10.1097/01.prs.0000299284.92862.6c. PMID 18317146. S2CID 21493967.
- ↑ Carpenter G (1909). "Case of acrocephaly with other congenital malformations". Proceedings of the Royal Society of Medicine. 2 (Sect Study Dis Child): 45–53, 199–201. doi:10.1177/003591570900201418. PMC 2047261. PMID 19974019.
- ↑ Beighton, Peter; Beighton, Greta (2012-12-06). The Man Behind the Syndrome. Springer Science & Business Media. p. 25. ISBN 9781447114154. Archived from the original on 2021-08-28. Retrieved 7 August 2018.
- ↑ "Carpenter syndrome". Genetic and Rare Diseases Information Center. National Center for Advancing Transnational Sciences. Archived from the original on 2021-01-25. Retrieved 2021-02-19.
- ↑ Frias, Jl; Felman, Ah; Rosenbloom, Al; Finkelstein, Sn; Hoyt, Wf; Hall, Bd (1978). "Normal intelligence in two children with Carpenter syndrome". American Journal of Medical Genetics. 2 (2): 191–9. doi:10.1002/ajmg.1320020210. PMID 263437.
- ↑ Jenkins, D; Seelow, D; Jehee, Fs; Perlyn, Ca; Alonso, Lg; Bueno, Df; Donnai, D; Josifova, D; Mathijssen, Im; Morton, Je; Orstavik, Kh; Sweeney, E; Wall, Sa; Marsh, Jl; Nurnberg, P; Passos-Bueno, Mr; Wilkie, Ao (June 2007). "RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity". American Journal of Human Genetics. 80 (6): 1162–70. doi:10.1086/518047. PMC 1867103. PMID 17503333.
- ↑ Twigg, SR; Lloyd, D; Jenkins, D; Elçioglu, NE; Cooper, CD; Al-Sannaa, N; Annagür, A; Gillessen-Kaesbach, G; Hüning, I; Knight, SJ; Goodship, JA; Keavney, BD; Beales, PL; Gileadi, O; McGowan, SJ; Wilkie, AO (Nov 2, 2012). "Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization". American Journal of Human Genetics. 91 (5): 897–905. doi:10.1016/j.ajhg.2012.08.027. PMC 3487118. PMID 23063620.
- 1 2 Paul A. Johnson, 2002
- ↑ Carpenter Syndrom-What is it?, 2007
External links
| Classification | |
|---|---|
| External resources |
|