X-linked lymphoproliferative disease
| X-linked lymphoproliferative disease | |
|---|---|
| Other names: Duncan disease, Purtilo syndrome | |
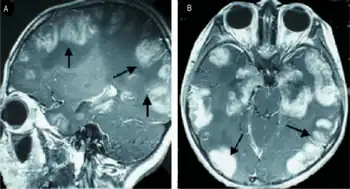 | |
| a,b)MRI scans of the brain show multiple non-homogeneous, hypodense masses along the bilateral cortex | |
| Symptoms | Reduced resistance to the Epstein-Barr virus (EBV), leading to infectious mononucleosis, hemophagocytic lymphohistiocytosis (HLH), dysgammaglobulinemia, non-Hodgkin lymphoma, aplastic anemia, vasculitis, chronic gastritis, skin lesions |
| Duration | Life long |
| Causes | Genetic (X-linked recessive) |
| Treatment | Chemotherapy, stem cell transplants |
| Frequency | 1 in 1,000,000 males (XLP1); 1 in 5,000,000 males (XLP2) |
X-linked lymphoproliferative disease (also known as Duncan disease[1]: 86 or Purtilo syndrome[2] and abbreviated as XLP[3]) is a lymphoproliferative disorder,[4] usually caused by SH2DIA gene mutations in males. XLP-positive individuals experience immune system deficiencies that render them unable to effectively respond to the Epstein-Barr virus (EBV),[3] a common virus in humans that typically induces mild symptoms or infectious mononucleosis (IM) in patients.[5] There are two currently known variations of the disorder, known as XLP1 (XLP Type 1) and XLP2. XLP1 is estimated to occur in approximately one in every million males, while XLP2 is rarer, estimated to occur in one of every five million males.[6] Due to therapies such as chemotherapy and stem cell transplants, the survival rate of XLP1 has increased dramatically since its discovery in the 1970s.[3][7]
Signs and symptoms
In boys with X-linked lymphoproliferative disorder, the inability to mount an immune response to EBV may lead to death via hemophagocytic lymphohistiocytosis (HLH). Patients may also develop dysgammaglobulinemia and malignant non-Hodgkin lymphoma, even without exposure to EBV. Other observed symptoms of XLP include aplastic anemia, vasculitis, chronic gastritis, and skin lesions, as well as IM.[3][8]
Nearly half of XLP patients express humoral immune anomalies, which can include diminished responses to vaccines and low levels of immunoglobulin G (IgG).[3][9]
Patients produce insufficient numbers of CD27 memory B cells.[10]
Cause
XLP1
XLP1 is caused by mutations in the SH2D1A gene, which is located at position Xq25 on the X-chromosome. This gene codes for an SH2 domain on a signal transducing protein called signaling lymphocyte activation molecule (SLAM)-associated protein, or SAP. A variety of mutations have been implicated in XLP1 expression, including deletions, single nucleotide changes, and incorrect splicing, although a correlation between the type of mutation and the severity of the disorder has not been established.[8]
These defects in SAP fundamentally change the function of two SLAM receptors, 2B4 (CD244) and NTB-A (SLAMF6). Typically, after the receptors bind to their associated ligands, the immunoreceptor tyrosine-based switch motifs (ITSMs) in their cytoplasms are phosphorylated, which activates cell-activating signaling pathways. In an XLP patient, the defects in SAP cause these receptors to induce an inhibitory, rather than activating effect. Ligand binding thus fails to activate Natural Killer (NK) and Cytotoxic T cells that typically eliminate EBV infection, leading to cytokine overproduction and tissue damage.[8]
The term "SH2" domain stands for src-homology 2 domain, which is a three-dimensional domain structure of about 100 amino acid residues. These domains are present in many signalling proteins because they permit specific, non-covalent bonding to proteins that contain phosphotyrosines. The amino acid residues adjacent to the phosphotyrosine on the target protein are what determine the unique binding specificity.[11]
XLP2
Any instance of XLP caused by a mutation not in SHD21A is categorized as XLP2, although the variation is typically caused by mutations in the XIAP (X-linked inhibitor of apoptosis, also known as BIRC4) gene.[3][6] XLP2 patients express different features from those typically found in XLP1 patients, such as splenomegaly and colitis. This variation is closely associated with HLH, so much so that some sources recommend classifying this condition as "X-linked familial hemophagocytic lymphohistiocytosis" instead of X-linked lymphoproliferative disease.[3][12]
Mutations in XIAP inhibit the expression of the gene, which usually regulates the rate of lymphocyte apoptosis during an immune response. Nonfunctional XIAP is unable to prevent lymphocytes from undergoing apoptosis in response to stimuli, which include the formation of the T-cell receptor (TCR)-CD3 complex, the binding the CD95 death receptor, and the activation TNF-associated apoptosis-inducing ligand receptor (TRAIL-R). This leads to higher rates of lymphocyte apoptosis during a normal immune response.[13]
XIAP-deficient individuals also produce low numbers of natural killer cells, a feature shared with XLP1 patients, which leads to a similarly inefficient response to EBV infection.[8][13]
Diagnosis
In terms of the evaluation of X-linked lymphoproliferative the following is done:[14]
- Blood test
- Genetic test
Treatment
Chemotherapy and hematopoietic stem-cell transplantation (HSCT) therapies have shown great success in treating XLP. The development of the two therapies, alongside more efficient monitoring techniques and supportive care, has reduced the overall mortality of the disease from 75% to 29%. Care differs depending on the phenotype of XLP expressed, with treatments varying between those experiencing HLH or lymphoma, and whether or not they have been infected with EBV.[3]
Still, a bone marrow transplant that includes CD34+ hematopoietic stem cells is the only known treatment for the disorder as a whole. Patients who cannot find a bone marrow donor have a survival rate of less than 20%. In addition to the typical restrictions imposed on donor-recipient matches, XLP1 patients who have been infected with EBV typically receive transplants from EBV-positive donors.[3]
Eponym
XLP is also known as Duncan Disease, after 6 of 18 males in the Duncan family died of lymphoproliferative disease, including fulminant infectious mononucleosis and lymphoma.[15] It is also called "Purtilo's Syndrome", after Dr. David Theodore Purtilo (1939–1992), a pioneering Pathologist and Immunologist at the American Army Center for Pathology in Washington, who discovered it in the early 1970s. A native of Duluth, Minnesota, he pioneered the research for this condition after discovering it in one of his patients. In the late 1980s, he resided in Omaha, Nebraska and died on September 28, 1992, in Florida, following a stroke before he could deliver a speech to a forum.[16]
References
- ↑ James WD, Berger TG, Elston DM (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ↑ "X-linked lymphoproliferative disease". MedlinePlus. U.S. National Library of Medicine. Archived from the original on 2020-09-19. Retrieved 2022-06-20.
- 1 2 3 4 5 6 7 8 9 Panchal N, Booth C, Cannons JL, Schwartzberg PL (April 2018). "X-Linked Lymphoproliferative Disease Type 1: A Clinical and Molecular Perspective". Frontiers in Immunology. 9: 666. doi:10.3389/fimmu.2018.00666. PMC 5893764. PMID 29670631.
- ↑ Rapini RP, Bolognia JL, Jorizzo JL (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. p. 808. ISBN 978-1-4160-2999-1.
- ↑ "About Epstein-Barr Virus (EBV)". U.S. Centers for Disease Control and Prevention. 2021-03-17. Archived from the original on 2016-08-08. Retrieved 2022-03-09.
- 1 2 "X-linked lymphoproliferative disease: MedlinePlus Genetics". MedlinePlus Genetics. U.S. National Library of Medicine. Archived from the original on 2022-03-24. Retrieved 2022-03-09.
- ↑ Sumegi J, Seemayer TA, Huang D, Davis JR, Morra M, Gross TG, et al. (June 2002). "A spectrum of mutations in SH2D1A that causes X-linked lymphoproliferative disease and other Epstein-Barr virus-associated illnesses". Leukemia & Lymphoma. 43 (6): 1189–1201. doi:10.1080/10428190290026240. PMID 12152986. S2CID 44576367.
- 1 2 3 4 Pende D, Meazza R, Marcenaro S, Aricò M, Bottino C (July 2019). "2B4 dysfunction in XLP1 NK cells: More than inability to control EBV infection". Clinical Immunology. 204: 31–36. doi:10.1016/j.clim.2018.10.022. PMID 30391652. S2CID 53220284.
- ↑ Panchal N, Booth C, Cannons JL, Schwartzberg PL (2018-04-04). "X-Linked Lymphoproliferative Disease Type 1: A Clinical and Molecular Perspective". Frontiers in Immunology. 9: 666. doi:10.3389/fimmu.2018.00666. PMC 5893764. PMID 29670631.
- ↑ Ma CS, Pittaluga S, Avery DT, Hare NJ, Maric I, Klion AD, et al. (February 2006). "Selective generation of functional somatically mutated IgM+CD27+, but not Ig isotype-switched, memory B cells in X-linked lymphoproliferative disease". The Journal of Clinical Investigation. 116 (2): 322–333. doi:10.1172/JCI25720. PMC 1332028. PMID 16424938.
- ↑ Abbas AK, Lichtman AH (2005). Cellular and Molecular Immunology (5th ed.). Philadelphia: Elsevier Saunders. ISBN 978-0-7216-0008-6.
- ↑ Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, et al. (August 2010). "XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease". Blood. 116 (7): 1079–1082. doi:10.1182/blood-2010-01-256099. PMC 2938130. PMID 20489057.
- 1 2 Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. (November 2006). "XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome". Nature. 444 (7115): 110–114. Bibcode:2006Natur.444..110R. doi:10.1038/nature05257. PMID 17080092. S2CID 4416976.
- ↑ "X-Linked Lymphoproliferative Disease (XLP) | NIH: National Institute of Allergy and Infectious Diseases". www.niaid.nih.gov. Archived from the original on 18 August 2022. Retrieved 22 November 2022.
- ↑ Purtilo DT, Cassel C, Yang JP (October 1974). "Letter: Fatal infectious mononucleosis in familial lymphohistiocytosis". The New England Journal of Medicine. 291 (14): 736. doi:10.1056/nejm197410032911415. PMID 4852784.
- ↑ Saxon W (3 October 1992). "David T. Purtilo, 53, a Specialist In Disorders of the Immune System". The New York Times. Archived from the original on 16 January 2018. Retrieved 20 June 2022.
External links
- GeneReview/NIH/UW entry on Lymphoproliferative Disease, X-Linked Archived 2022-01-20 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|