Mohr–Tranebjærg syndrome
| Mohr–Tranebjærg syndrome | |
|---|---|
| Other names | Deafness–dystonia–optic neuronopathy syndrome, Deafness–dystonia–optic atrophy syndrome, Deafness syndrome, progressive, with blindness, dystonia, fractures, and mental deficiency |
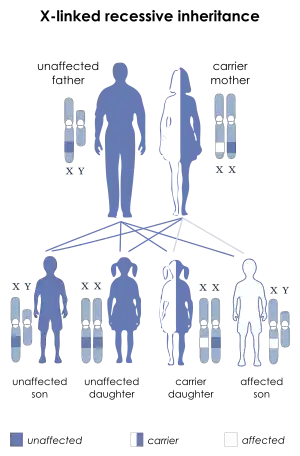 | |
| Mohr–Tranebjærg syndrome is inherited in an X-linked recessive manner | |
Mohr–Tranebjærg syndrome (MTS) is a rare X-linked recessive syndrome also known as deafness–dystonia syndrome and caused by mutation in the TIMM8A gene. It is characterized by clinical manifestations commencing with early childhood onset hearing loss, followed by adolescent onset progressive dystonia or ataxia, visual impairment from early adulthood onwards and dementia from the 4th decade onwards.[1] The severity of the symptoms may vary, but they progress usually to severe deafness and dystonia and sometimes are accompanied by cortical deterioration of vision and mental deterioration.
Signs and Symptoms
Mohr-Tranebjærg is characterized by a variety of symptoms affecting hearing loss. In the span of a subject with Mohr-Tranebjærg, by the age of early childhood, around 18 months, the development of quickly progressive prelingual or postlingual sensorineural hearing loss.[2] The presence of auditory symptoms is characterized by preserved oto-acoustic emissions, abnormal auditory brain stem response, poor speech, and worsening in auditory symptoms even with auditory devices.[2] Also, neuropsychological manifestations, possibly consisting of personality changes, paranoia, and mild intellectual deficit could emerge.[2] During adolescence, there is a possibility that a slowly progressive movement disorder, similar to gegenhalten, dystonia, or ataxia could develop.[2] Patients with Mohr-Tranebjærg experience reduced visual acuity, photophobia, acquired color vision defect and central scotomas developing at around 20 years of age, and leading to more severe blindness by the age of 30-40.[2]
The first symptom of DDON syndrome is hearing loss caused by nerve damage in the inner ear (sensorineural hearing loss), which begins in early childhood. The hearing impairment worsens over time, and most affected individuals have profound hearing loss by age 10.[3]
Individuals with Mohr Tranebjærg syndrome have normal vision during childhood, but they may develop vision problems due to breakdown of the nerves that carry information from the eyes to the brain (optic atrophy). Affected individuals can develop an increased sensitivity to light (photophobia) or other vision problems beginning in adolescence. Their sharpness of vision (visual acuity) slowly worsens, often leading to legal blindness in mid-adulthood.[3]
Genetics
This condition is caused by mutations in the TIMM8A gene. This gene is located on the long arm of X chromosome (Xq22).
The protein encoded by this gene localizes to the intermembrane space in mitochondria where it functions in the import of nuclear encoded proteins into the mitochondrial inner membrane. How this produces the clinical picture is not yet clear.
The pattern of inheritance is X-linked recessive. Where the female is a carrier, male offspring have a 50 percent chance of inheriting the disease and female offspring have a 50 percent chance of being a carrier. Where the male is affected, male offspring do not inherit the pathogenic mutation and females are obligate carriers.[4]
TIMM8A is also referred to as DDP. Koehler determined the function of the DDP gene and concluded that the Mohr-Tranebjaerg syndrome is a novel type of mitochondrial disease that is most likely caused by a defective mitochondrial protein-import system. [5]
Diagnosis
A combination of hearing impairment and recurrent infections due to XLA in a male patient should elicit sequencing of the TIMM8A gene. Neuroimaging is employed to verify the presence of cerebral atrophy. In cases of suspected CGS; testing for XLA is possible.[6]
Differential diagnosis
This is long and includes
- Arts syndrome
- Autosomal recessive nonsyndromic sensorineural deafness type DFNB
- Friedreich ataxia
- MELAS syndrome
- Mitochondrial DNA depletion syndrome (encephalomyopathic form with methylmalonic aciduria)
- McLeod neuroacanthocytosis syndrome
- Pendred syndrome
- Usher syndrome type 1 and 2
- Wolfram syndrome
- X-linked spinocerebellar ataxia type 3 and 4
Treatment
Symptomatic Treatment
The treatment for Mohr-Tranebjærg vary depending on the symptoms and the evolution of these symptoms over time.[7] Since hearing loss is prevalent in those with Mohr-Tranebjærg, hearing aids, devices, or cochlear implants are considered for increasingly serious cases.
Immunoglobin
In those patients with secondary complications from the syndrome, intravenous immunoglobin may act to prevent infections in X-linked agammaglobulinemia, an inherited immune system disorder that can reduce the ability to fight infections. Similarly, those with XLA should avoid live viral vaccines.[7] Patients that reach adulthood should seek regular evaluations from a neurologist to test the advancement of possible dementia and psychiatric manifestations[7]
Genetic Counseling
Since the pattern of inheritance for Mohr-Tranebjærg is known to be X-linked recessive, genetic counseling is considered beneficial to families with known carriers.[7] In the case that a female is a carrier, the statistical risk of male offspring inheriting the disease is 50%, while the probability that a female offspring will be a carrier is 50%.[7] However, when a male is the carrier, the probability of having a female carrier offspring is 100%, and male offspring will not have any chance of inheriting the syndrome.[7] Because of the statistical probabilities that each sex inherits the syndrome, males are affected by Mohr-Tranebjærg more frequently than females are.[6] Females who carry one copy of the TIMM8A gene (described in Genetics) are usually unaffected, however, may develop mild hearing loss and dystonia.[8]
Prognosis
Prognosis is poor. The combination of deafness and blindness severely affects communication, while the ongoing movement disorder results in an increasingly unstable gait. Life expectancy is highly variable and can range from death in the teenage years (after a rapidly progressive dystonia) to those that live into their 60's.[1]
History
This condition was first described in 1960.[9]
Epidemiology
Mohr-Tranebjᴂrg syndrome (MTS) prevalence is unknown. More than 90 cases (in 37 families) are known, but not all cases have been reported in the literature.[10]
See also
- Mitochondrial disorders
- TIMM13 and TIMM8A
- Dystonia
References
- 1 2 Makar, A. B.; McMartin, K. E.; Palese, M.; Tephly, T. R. (June 1975). "Formate assay in body fluids: application in methanol poisoning". Biochemical Medicine. 13 (2): 117–126. doi:10.1016/0006-2944(75)90147-7. ISSN 0006-2944. PMID 1.
- 1 2 3 4 5 Hendrickson, W. A.; Ward, K. B. (1975-10-27). "Atomic models for the polypeptide backbones of myohemerythrin and hemerythrin". Biochemical and Biophysical Research Communications. 66 (4): 1349–1356. doi:10.1016/0006-291x(75)90508-2. ISSN 1090-2104. PMID 5.
- 1 2 Wiesmann, U. N.; DiDonato, S.; Herschkowitz, N. N. (1975-10-27). "Effect of chloroquine on cultured fibroblasts: release of lysosomal hydrolases and inhibition of their uptake". Biochemical and Biophysical Research Communications. 66 (4): 1338–1343. doi:10.1016/0006-291x(75)90506-9. ISSN 1090-2104. PMID 4.
- ↑ Hendrickson, W. A.; Ward, K. B. (1975-10-27). "Atomic models for the polypeptide backbones of myohemerythrin and hemerythrin". Biochemical and Biophysical Research Communications. 66 (4): 1349–1356. doi:10.1016/0006-291x(75)90508-2. ISSN 1090-2104. PMID 5.
- ↑ "OMIM Entry #304700 - MOHR-TRANEBJAERG SYNDROME; MTS". OMIM - Online Mendelian Inheritance in Man. Retrieved 2021-04-26.
{{cite web}}: CS1 maint: url-status (link) - 1 2 "Mohr-Tranebjaerg syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2021-04-26.
- 1 2 3 4 5 6 RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Mohr Tranebjaerg syndrome". www.orpha.net. Retrieved 2021-04-26.
- ↑ Wiesmann, U. N.; DiDonato, S.; Herschkowitz, N. N. (1975-10-27). "Effect of chloroquine on cultured fibroblasts: release of lysosomal hydrolases and inhibition of their uptake". Biochemical and Biophysical Research Communications. 66 (4): 1338–1343. doi:10.1016/0006-291x(75)90506-9. ISSN 1090-2104. PMID 4.
- ↑ Mohr J, Mageroy K (1960). "Sex-linked deafness of a possibly new type". Acta Genet Stat Med. 10 (1–3): 54–62. doi:10.1159/000151118. PMID 13771732.
- ↑ Makar, A. B.; McMartin, K. E.; Palese, M.; Tephly, T. R. (June 1975). "Formate assay in body fluids: application in methanol poisoning". Biochemical Medicine. 13 (2): 117–126. doi:10.1016/0006-2944(75)90147-7. ISSN 0006-2944. PMID 1.
External links
- GeneReviews/NCBI/NIH/UW entry on Deafness–Dystonia–Optic Neuronopathy Syndrome
- MTS — a page at NIH website