Haemophilia B
| Haemophilia B | |
|---|---|
| Other names: Hemophilia B; Christmas disease; factor IX deficiency; FIX deficiency;[1] royal disease[2] | |
.svg.png.webp) | |
| This condition is inherited in an X-linked recessive manner. | |
| Specialty | Haematology |
| Symptoms | Easy bruising and bleeding[3] |
| Complications | Bleeding into joints or brain[3] |
| Types | Mild, moderate, severe[2] |
| Causes | Genetic disorder (X-linked recessive)[3] |
| Diagnostic method | Blood tests (aPTT, factor IX activity level)[2] |
| Differential diagnosis | Hemophilia A, hemophilia C, Von Willebrand disease[2] |
| Treatment | Factor IX concentrate[1][2] |
| Prognosis | Often decreased life expectancy[4] |
| Frequency | 1 in 25,000 male newborns[2] |
Haemophilia B, also known as Christmas disease, is a bleeding disorder that occurs due to a genetic mutation.[3] Symptoms include easy bruising and bleeding.[3] Complications can include bleeding into joints or the brain.[3] Depending on the severity, the disease may become apparent between the newborn period and adulthood.[2]
It is generally due to an inherited mutation, which occurs in an X-linked recessive pattern.[3] About a third of cases occur due to a new mutation.[1] Rarely it may occur later in life due to the production of antibodies against factor IX.[2] The underlying mechanism involves a lack of factor IX, a part of the coagulation cascade.[1] Diagnosis may be suspected based on a blood test for aPTT and confirmed by a factor IX activity level.[2] It is divided into mild, moderate, and severe depending on a person's factor IX levels.[2]
Treatment is with factor IX concentrate, with recombinant factor IX products often recommended.[1] These may be used preventatively or when bleeding occurs.[1] Fresh frozen plasma may be used if this is not available.[2] Aminocaproic acid may also be used.[1] Gene therapy, in the form of etranacogene dezaparvovec, became an option in the United States in 2022.[2] Despite availability of treatment life expectancy is reduced by about 25%;[4] though, without treatment many died around the age of 12.[5]
Haemophilia B affects about 1 in 25,000 male newborns.[2] Males are primarily affected; though, a few females may also have the disease.[2] It is about 5 times less common than haemophilia A.[6] It was first recognized as a distinct disease in 1952.[2] Many members of European royal families were affected as Queen Victoria carried the mutation.[2]
Signs and symptoms
Symptoms include easy bruising, urinary tract bleeding (haematuria), nosebleeds (epistaxis), and bleeding into joints (haemarthrosis).
Complications
People with bleeding disorders show a higher incidence of periodontal disease as well as dental caries, concerning the fear of bleeding which leads to a lack of oral hygiene and oral health care. The most prominent oral manifestation of a mild haemophilia B would be gingival bleeding during exfoliation of primary dentition, or prolonged bleeding after an invasive procedure/tooth extraction; In severe haemophilia, there may be spontaneous bleeding from the oral tissues (e.g. soft palate, tongue, buccal mucosa), lips and gingiva, with ecchymoses. In rare cases, haemarthrosis (bleeding into joint space) of the temporomandibular joint (TMJ) may be observed.[7]
People with haemophilia will experience many episodes of oral bleeding over their lifetime. Average 29.1 bleeding events per year are serious enough to require factor replacement in F VIII-deficient people which 9% involved oral structures. Children with severe haemophilia have significant lower prevalence of dental caries and lower plaque scores compared with matched, healthy controls.[8]
Genetics
The factor IX gene is located on the X chromosome (Xq27.1-q27.2). It is an X-linked recessive trait, which explains why males are affected in greater numbers.[9][10]
In 1990, George Brownlee and Merlin Crossley showed that two sets of genetic mutations were preventing two key proteins from attaching to the DNA of people with a rare and unusual form of haemophilia B – haemophilia B Leyden – where sufferers experience episodes of excessive bleeding in childhood but have few bleeding problems after puberty.[10] This lack of protein attachment to the DNA was thereby turning off the gene that produces clotting factor IX, which prevents excessive bleeding.[10]
Pathophysiology
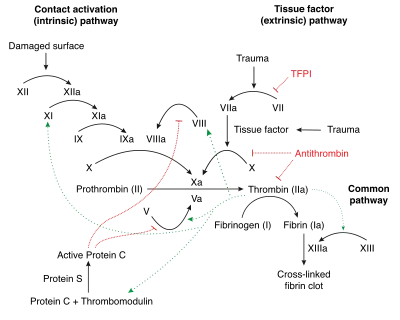
Factor IX deficiency leads to an increased propensity for haemorrhage, which can be either spontaneously or in response to mild trauma.[11]
Factor IX deficiency can cause interference of the coagulation cascade, thereby causing spontaneous haemorrhage when there is trauma. Factor IX when activated activates factor X which helps fibrinogen to fibrin conversion.[11]
Factor IX becomes active eventually in coagulation by cofactor factor VIII (specifically IXa). Platelets provide a binding site for both cofactors. This complex (in the coagulation pathway) will eventually activate factor X.[12]
Diagnosis

The diagnosis for haemophilia B can be done via the following tests/methods:[13]
- Coagulation screening test
- Bleeding scores
- Coagulation factor assays
Differential diagnosis
The differential diagnosis for this inherited condition is the following: haemophilia A, factor XI deficiency, von Willebrand disease, fibrinogen disorders and Bernard–Soulier syndrome[10]
Treatment
Treatment may be given preventatively or intermittently with bleeding. It includes intravenous infusion of factor IX and/or blood transfusions. NSAIDS should be avoided once the diagnosis is made since they can exacerbate a bleeding episode. Any surgical procedure should be done with concomitant tranexamic acid.[6][14]
Teeth
Surgical treatment, including a simple dental extraction, must be planned to minimize the risk of bleeding, excessive bruising, or haematoma formation. Soft vacuum-formed splints can be used to provide local protection following a dental extraction or prolonged post-extraction bleed.[15]
History
In the 1950s and 1960s, with newfound technology and gradual advances in medicine, pharmaceutical scientists found a way to take the factor IX from fresh frozen plasma (FFP) and give it to those with haemophilia B. Though they found a way to treat the disease, the FFP contained only a small amount of factor IX, requiring large amounts of FFP to treat an actual bleeding episode, which resulted in the person requiring hospitalization. By the mid-1960s scientists found a way to get a larger amount of factor IX from FFP. By the late 1960s, pharmaceutical scientists found methods to separate the factor IX from plasma, which allows for neatly packaged bottles of factor IX concentrates. With the rise of factor IX concentrates it became easier for people to get treatment at home.[16] Although these advances in medicine had a significant positive impact on the treatment of haemophilia, there were many complications that came with it. By the early 1980s, scientists discovered that the medicines they had created were transferring blood-borne viruses, such as hepatitis, and HIV, the virus that causes AIDS. With the rise of these deadly viruses, scientists had to find improved methods for screening the blood products they received from donors. In 1982, scientists made a breakthrough in medicine and were able to clone factor IX gene. With this new development it decreased the risk of the many viruses. Although the new factor was created, it wasn't available for haemophilia B until 1997.
Terminology
It is also known by the eponym Christmas disease,[17] named after Stephen Christmas, the first person described with haemophilia B. In addition, the first report of its identification was published in the Christmas edition of the British Medical Journal.[6][18]
Society and culture
In 2009, an analysis of genetic markers revealed that haemophilia B was the blood disease affecting many European royal families of Great Britain, Germany, Russia and Spain: so-called "Royal Disease".[19][20]
See also
- Haemophilia C
- Haemophilia in European royalty
References
- 1 2 3 4 5 6 7 "Hemophilia B | NBDF". National Hemophilia Foundation. Archived from the original on 7 June 2023. Retrieved 13 September 2023.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 "Hemophilia B". NORD. 2023. Archived from the original on 31 May 2023. Retrieved 14 September 2023.
- 1 2 3 4 5 6 7 "Hemophilia B - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Archived from the original on 23 November 2022. Retrieved 13 September 2023.
- 1 2 "Estimating hemophilia prevalence and life expectancy | Canadian Blood Services". www.blood.ca. Archived from the original on 5 July 2022. Retrieved 14 September 2023.
- ↑ Alshaikhli, Alfarooq; Rokkam, Venkata R. (2023). "Hemophilia B". StatPearls. StatPearls Publishing. Archived from the original on 7 December 2022. Retrieved 14 September 2023.
- 1 2 3 "Haemophilia B (Factor IX Deficiency) information | Patient". Patient. Archived from the original on 2016-04-08. Retrieved 2016-04-21.
- ↑ "Hemophilia A" (PDF). College of Dental Hygienists of Ontario. September 2, 2015.
- ↑ Glick, Michael (2015). Burket's Oral Medicine. USA: People's Medical Publishing House. pp. 473, 475, 481, 482. ISBN 978-1-60795-188-9.
- ↑ "OMIM Entry - # 306900 - HEMOPHILIA B; HEMB". omim.org. Archived from the original on 2017-01-28. Retrieved 2016-10-07.
- 1 2 3 4 "Hemophilia". Archived from the original on 2018-07-28. Retrieved 2021-09-28.
- 1 2 "Hemophilia B: Practice Essentials, Background, Pathophysiology". eMedicine. Medscape. 24 August 2016. Archived from the original on 10 October 2016. Retrieved 7 October 2016.
- ↑ "Factor IX Deficiency: Background, Pathophysiology, Epidemiology". eMedicine. Medscape. 24 August 2016. Archived from the original on 6 October 2016. Retrieved 7 October 2016.
- ↑ Konkle, Barbara A.; Josephson, Neil C.; Nakaya Fletcher, Shelley (1 January 1993). "Hemophilia B". GeneReviews. Archived from the original on 20 October 2019. Retrieved 7 October 2016.update 2014
- ↑ Beck, Norman (2009). Diagnostic hematology. London: Springer. p. 416. ISBN 9781848002951. Archived from the original on 14 October 2021. Retrieved 7 October 2016.
- ↑ Andrew Brewer, Maria Elvira Correa (May 2006). "Guildelines for Dental Treatment of Patients with Inherited Bleeding Disorders" (PDF). Treatment of Hemophilia. 40: 9 – via World Federation of Hemophilia (WFH).
- ↑ Schramm, Wolfgang (November 2014). "The history of haemophilia – a short review". Thrombosis Research. 134: S4–S9. doi:10.1016/j.thromres.2013.10.020. ISSN 1879-2472. PMID 24513149. – via ScienceDirect (Subscription may be required or content may be available in libraries.)
- ↑ "Hemophilia B: MedlinePlus Medical Encyclopedia". medlineplus.gov. Archived from the original on 2016-07-05. Retrieved 2016-09-21.
- ↑ Biggs R, Douglas AS, MacFarlane RG, Dacie JV, Pitney WR, Merskey C, O'Brien JR (1952). "Christmas disease: a condition previously mistaken for haemophilia". Br Med J. 2 (4799): 1378–82. doi:10.1136/bmj.2.4799.1378. PMC 2022306. PMID 12997790.
- ↑ Michael Price (8 October 2009). "Case Closed: Famous Royals Suffered From Hemophilia". ScienceNOW Daily News. AAAS. Archived from the original on 20 October 2013. Retrieved 9 October 2009.
- ↑ Evgeny I. Rogaev; et al. (8 October 2009). "Genotype Analysis Identifies the Cause of the "Royal Disease"". Science. 326 (5954): 817. Bibcode:2009Sci...326..817R. doi:10.1126/science.1180660. PMID 19815722. S2CID 206522975.subscription required
Further reading
- Franchini, Massimo; Frattini, Francesco; Crestani, Silvia; Sissa, Cinzia; Bonfanti, Carlo (1 January 2013). "Treatment of hemophilia B: focus on recombinant factor IX". Biologics: Targets and Therapy. 7: 33–38. doi:10.2147/BTT.S31582. ISSN 1177-5475. PMC 3575125. PMID 23430394.
- Nathwani, Amit C.; Reiss, Ulreke M.; Tuddenham, Edward G.D.; Rosales, Cecilia; Chowdary, Pratima; McIntosh, Jenny; Della Peruta, Marco; Lheriteau, Elsa; Patel, Nishal; Raj, Deepak; Riddell, Anne; Pie, Jun; Rangarajan, Savita; Bevan, David; Recht, Michael; Shen, Yu-Min; Halka, Kathleen G.; Basner-Tschakarjan, Etiena; Mingozzi, Federico; High, Katherine A.; Allay, James; Kay, Mark A.; Ng, Catherine Y.C.; Zhou, Junfang; Cancio, Maria; Morton, Christopher L.; Gray, John T.; Srivastava, Deokumar; Nienhuis, Arthur W.; Davidoff, Andrew M. (20 November 2014). "Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B". New England Journal of Medicine. 371 (21): 1994–2004. doi:10.1056/NEJMoa1407309. ISSN 0028-4793. PMC 4278802. PMID 25409372.
External links
| Classification | |
|---|---|
| External resources |