Barth syndrome
| Barth Syndrome | |
|---|---|
| Other names: 3-Methylglutaconic aciduria type II, | |
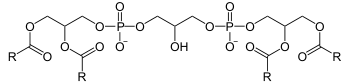 | |
| Cardiolipin | |
Barth syndrome (BTHS) is an X-linked[1] genetic disorder. The disorder, which affects multiple body systems, is diagnosed almost exclusively in males. Barth's syndrome (BTHS), or cardioskeletal myopathy, also known as type II 3-methylglutaconium aciduria, is a congenital syndrome of phospholipid metabolism characterized by increased and weakened cardiomyopathy, being inherited from the mother, the syndrome modifies the BTHS gene on the chromosome. X. It is a rare and serious disorder that affects only men, and sometimes fatal.
The syndrome is named after neurologist Dr. Peter Barth, a pediatric neurologist, who through his studies around 1983, described a class scheme demonstrating that the disease is not communicable. There is still no cure for Barth Syndrome, but there are ways to stabilize some symptoms of the disease.[2]
It is caused by mutations in the TAZ gene, which encodes the enzymatic protein tafazzin. The TAZ gene is located on the X chromosome, which explains the fact that the syndrome's inheritance is linked to transmission through this chromosome, thus being inherited from the mother.
Synonyms,AGM2,Type 2 3-methylglutaconic aciduria,BTHS,Cardioskeletal myopathy - neutropenia
Signs and symptoms
Though not always present, the cardinal characteristics of this multi-system disorder include: cardiomyopathy (dilated or hypertrophic, possibly with left ventricular noncompaction and/or endocardial fibroelastosis),[3][4] neutropenia (chronic, cyclic, or intermittent),[4] underdeveloped skeletal musculature and muscle weakness,[5] growth delay,[4] exercise intolerance, cardiolipin abnormalities,[6][7] and 3-methylglutaconic aciduria.[4] It can be associated with stillbirth.[8]
Barth syndrome is manifested in a variety of ways at birth. A majority of BTHS patients are hypotonic at birth, show signs of cardiomyopathy within the first few months of life, and experience a deceleration in growth in the first year, despite adequate nutrition. As patients progress into childhood, their height and weight lag significantly behind other children. While most patients express normal intelligence, a high proportion of BTHS patients also express mild or moderate learning disabilities. Physical activity is also hindered due to diminished muscular development and muscular hypotonia. Many of these disorders are resolved after puberty. Growth accelerates during puberty, and many patients reach a normal adult height.[9]
Cardiomyopathy is one of the more severe manifestations of BTHS. The myocardium is dilated, reducing the systolic pump of the ventricles. For this reason, most BTHS patients have left myocardial thickening (hypertrophy). While cardiomyopathy can be life-threatening, it is commonly resolved or substantially improved in BTHS patients after puberty.[9] Neutropenia is another deadly manifestation of BTHS. Neutropenia is a granulocyte disorder that results in a low production of neutrophils, the body's primary defenders against bacterial infections. Surprisingly, however, BTHS patients have relatively fewer bacterial infections than other patients with neutropenia.[10]
Clinical description
The clinical manifestations presented by patients with this syndrome include: dilated cardiomyopathy (enlarged and weakened heart). Most men diagnosed with BTHS present DCM up to ten years of age, and endocardial fibroelastosis (EFE) or left ventricular non-compaction (LVNV) may occur. When symptoms occur during pregnancy between the 2nd and 3rd trimester, it can cause complications to the fetus, such as heart failure, hydrops fetalis, miscarriage or stillbirth. In adolescents, the ventricular arrhythmia responsible for sudden cardiac death can occur.
Skeletal myopathy (muscle weakness) causes delay in motor development, loss of strength and muscle tone, exercise intolerance and lethargy. During the developmental phase, boys present deficits in weight height and even pubertal delay. With regard to neutropenia, some patients develop a chronic, cyclic or intermittent condition. These changes in neutrophils allow patients with the syndrome to be vulnerable to infections, such as mouth ulcers, skin abscesses, fever and bacterial infections.[3]
 Broad forehead, round face, prominent ears
Broad forehead, round face, prominent ears Neutropenia
Neutropenia
Cause
Mutations in the tafazzin gene (TAZ, also called G4.5) are closely associated with Barth syndrome. The tafazzin gene product functions as an acyltransferase in complex lipid metabolism.[6][7] In 2008, Dr. Kulik found that all the BTHS individuals that he tested had abnormalities in their cardiolipin molecules, a lipid found inside the mitochondria of cells.[11] Cardiolipin is intimately connected with the electron transport chain proteins and the membrane structure of the mitochondria which is the energy producing organelle of the cell. The human tafazzin gene, NG_009634, is listed as over 10,000 base pairs in length and the full-length mRNA, NM_000116, is 1919 nucleotides long encoding 11 exons with a predicted protein length of 292 amino acids and a molecular weight of 33.5 kDa. The tafazzin gene is located at Xq28;[12] the long arm of the X chromosome. Mutations in tafazzin that cause Barth syndrome span many different categories: missense, nonsense, deletion, frameshift, splicing (see Human Tafazzin (TAZ) Gene Mutation & Variation Database).[13]
iPLA2-VIA has been suggested as a target for treatment.[14]
Genetics
BTHS is an X-linked recessive inheritance condition, usually transmitted from mother to baby, where a mother carrying the Barth syndrome mutation (the gene is called tafazzin - also called TAZ or G4.5) does not has symptoms of this disorder, probably due to distorted X-chromosome inactivation.
There are some literatures that address cases of women with the syndrome who may not have developed the disease, but who have a 50% chance of transmitting the modified gene to their children. In these cases, there is a 50% chance that a child generated by a carrier of the syndrome will have a modified gene. The descendants of this affected man may be the carriers, usually asymptomatic, since the gene will be modified. Thus, Barth's cases in family histories must be investigated in order to define the genetic risk in each family. Any boy who is matrilineally related to an individual with Barth syndrome should be tested for this disease because the phenotype can vary widely, even among affected siblings.
It is understood that BTHS is caused by a mutation in the Tafazzin – TAZ gene. Tafazzin is known as a protein, in humans it is encoded by the TAZ gene. It is highly expressed in cardiac and skeletal muscle and acts as a phospholipid-lysophospholipid transacylase.
Gene-encoded protein and its function
BTHS is caused by mutations in the TAZ gene (tafazzin - protein; Xq28), which encodes the acyltransferase Taz1p involved in the metabolism of cardiolipin, a phospholipid that plays an important role in the inner membranes of mitochondria. Deficiency in Taz1p function results in abnormal cardiolipin remodeling that ends up compromising the structure and/or function of the mitochondrial respiratory chain.
Barth's syndrome alters the BTHS gene on the X chromosome, which can cause cardiac, immunological and male developmental complications. It can be seen by a professional at birth or within a few months after birth.
Diagnosis
Diagnosis may include blood, urinary, and/or imaging tests, as best discussed below:
- Blood tests used for white blood cell counts.
- Urinalysis used to determine the presence of an increase in organic acid characteristically seen with BTHS.
- Echocardiogram or cardiac ultrasound to assess the structure, function and condition of the heart.
- DNA sequencing analysis to determine BTHS gene abnormality.
Differential diagnosis
Differential diagnosis includes hereditary, dilated and nutritional cardiomyopathy and cyclic or idiopathic neutropenia.
Treatment
Currently there is no treatment for Barth syndrome, although some of the symptoms can be successfully managed. There are currently clinical trials happening for possible treatments in the future like AAV9-mediated TAZ gene replacement strategy. The University of Florida has run a research investigation exploring the TAZ gene that has shown initial promising results, but more research and pre-clinical and clinical testing needs to be done before the gene therapy is approved by the FDA as a treatment.[15][16]
Epidemiology
It has been documented, to date, in more than 120 males (see Human Tafazzin (TAZ) Gene Mutation & Variation Database).[13] It is believed to be severely under-diagnosed[17] and may be estimated to occur in 1 out of approximately 300,000 births. Family members of the Barth Syndrome Foundation and its affiliates live in the US, Canada, the UK, Europe, Japan, South Africa, Kuwait, and Australia.
Barth syndrome has been predominately diagnosed in males, although by 2012 a female case had been reported.[18]
Early diagnosis of the syndrome is complicated and critical. According to some international literature, it is believed that its prevalence is estimated at around 1 / 454,000 and an incidence at 1 / 140,000 (South West England, South Wales) to 1 / 300,000 - 1 / 400,000 live births (USA). In other materials, it addresses that the syndrome affects approximately one in every 200,000 births. In existing cases, the occurrence may be underdiagnosed due to its complex nature. Bringing to the Brazilian reality, there may be cases, however, in scientific literature there are few materials found.
History
The syndrome was named for Dr. Peter Barth (pediatric neurologist) (1932-) in the Netherlands for his research and discovery in 1983.[5] He described a pedigree chart, showing that this is an inherited trait.
See also
- 3-Methylglutaconic aciduria
- noncompaction cardiomyopathy: mutations to the affected genes in Barth syndrome are also present here.
References
- ↑ Claypool SM, Boontheung P, McCaffery JM, Loo JA, Koehler CM (December 2008). "The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome". Mol. Biol. Cell. 19 (12): 5143–55. doi:10.1091/mbc.E08-09-0896. PMC 2592642. PMID 18799610.
- ↑ Orpha.net. [Disponível em: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=111&lng=PT "Síndrome de Barth"]. Archived from the original on 2021-08-12. Retrieved 2021-10-25.
{{cite web}}: Check|url=value (help) - 1 2 Spencer CT, Bryant RM, Day J, et al. (August 2006). "Cardiac and clinical phenotype in Barth syndrome". Pediatrics. 118 (2): e337–46. doi:10.1542/peds.2005-2667. PMID 16847078. S2CID 23163528.
- 1 2 3 4 Kelley RI, Cheatham JP, Clark BJ, et al. (November 1991). "X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria". The Journal of Pediatrics. 119 (5): 738–47. doi:10.1016/S0022-3476(05)80289-6. PMID 1719174.
- 1 2 Barth PG, Scholte HR, Berden JA, et al. (December 1983). "An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes". Journal of the Neurological Sciences. 62 (1–3): 327–55. doi:10.1016/0022-510X(83)90209-5. PMID 6142097. S2CID 22790290.
- 1 2 Schlame M, Kelley RI, Feigenbaum A, et al. (December 2003). "Phospholipid abnormalities in children with Barth syndrome". Journal of the American College of Cardiology. 42 (11): 1994–9. doi:10.1016/j.jacc.2003.06.015. PMID 14662265.
- 1 2 Vreken P, Valianpour F, Nijtmans LG, et al. (December 2000). "Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome". Biochemical and Biophysical Research Communications. 279 (2): 378–82. doi:10.1006/bbrc.2000.3952. PMID 11118295.
- ↑ Steward CG, Newbury-Ecob RA, Hastings R, et al. (October 2010). "Barth syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth". Prenat. Diagn. 30 (10): 970–6. doi:10.1002/pd.2599. PMC 2995309. PMID 20812380.
- 1 2 Kelley RI, [cited 6 Dec 2011]. “Barth Syndrome - X-linked Cardiomyopathy and Neutropenia”. Department of Pediatrics, Johns Hopkins Medical Institutions. Available from: "Archived copy". Archived from the original on 2004-12-14. Retrieved 2004-12-19.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ Barth Syndrome Foundation, 28 Jun 2011. “Diagnosis of Barth Syndrome”. Available from: "Barth Syndrome Foundation : Home". Archived from the original on 2012-04-26. Retrieved 2011-12-06.
- ↑ Kulik W, van Lenthe H, Stet FS, et al. (February 2008). "Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome". Clinical Chemistry. 54 (2): 371–8. doi:10.1373/clinchem.2007.095711. PMID 18070816.
- ↑ Bione S, D'Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D (April 1996). "A novel X-linked gene, G4.5. is responsible for Barth syndrome". Nature Genetics. 12 (4): 385–9. doi:10.1038/ng0496-385. PMID 8630491. S2CID 23539265.
- 1 2 "Barth Syndrome Foundation : Home". Archived from the original on 2009-09-23. Retrieved 2009-04-17.
- ↑ Malhotra A, Edelman-Novemsky I, Xu Y, et al. (February 2009). "Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome". Proc. Natl. Acad. Sci. U.S.A. 106 (7): 2337–41. Bibcode:2009PNAS..106.2337M. doi:10.1073/pnas.0811224106. PMC 2650157. PMID 19164547.
- ↑ Suzuki-Hatano, Silveli; Saha, Madhurima; Rizzo, Skylar A.; Witko, Rachael L.; Gosiker, Bennett J.; Ramanathan, Manashwi; Soustek, Meghan S.; Jones, Michael D.; Kang, Peter B. (2018-08-02). "AAV-Mediated TAZ Gene Replacement Restores Mitochondrial and Cardioskeletal Function in Barth Syndrome". Human Gene Therapy. 30 (2): 139–154. doi:10.1089/hum.2018.020. ISSN 1043-0342. PMC 6383582. PMID 30070157.
- ↑ "Gene therapy for heart, skeletal muscle disorder shows promise in preclinical model | UF Health, University of Florida Health". 13 December 2018. Archived from the original on 13 April 2019. Retrieved 25 October 2021.
- ↑ Cantlay AM, Shokrollahi K, Allen JT, Lunt PW, Newbury-Ecob RA, Steward CG (September 1999). "Genetic analysis of the G4.5 gene in families with suspected Barth syndrome". The Journal of Pediatrics. 135 (3): 311–5. doi:10.1016/S0022-3476(99)70126-5. PMID 10484795.
- ↑ Cosson L, Toutain A, Simard G, Kulik W, Matyas G, Guichet A, Blasco H, Maakaroun-Vermesse Z, Vaillant MC, Le Caignec C, Chantepie A, Labarthe F (May 2012). "Barth syndrome in a female patient". Mol Genet Metab. 106 (1): 115–20. doi:10.1016/j.ymgme.2012.01.015. PMID 22410210.
External links
| Classification | |
|---|---|
| External resources |
|