Occipital horn syndrome
| Occipital horn syndrome | |
|---|---|
| Other names: Ehlers-Danlos syndrome type IX; X-Linked Cutis Laxa | |
.svg.png.webp) | |
| This condition is inherited in an X-linked recessive manner. | |
| Complications | Aortic aneurysms |
| Medication | Droxidopa, copper-histidine injections |
Occipital horn syndrome (OHS), formerly considered a variant of Ehlers–Danlos syndrome,[1] is an X-linked recessive mitochondrial and connective tissue disorder. It is caused by a deficiency in the transport of the essential mineral copper, associated with mutations in the ATP7A gene.[2][3]
Only about 2/3 of children with OHS are thought to have genetically inherited the disorder; the other 1/3 do not have the disease in their family history. Since the disorder is X-linked recessive the disease affects more males. This is because they do not have a second X chromosome, unlike females, so essentially are lacking the 'backup' copy with proper function. Females are much more likely to be carriers only. For a female to be affected they must carry two defective X chromosomes, not just one.[4]
The disorder is considered a milder variant of Menkes disease.[5]
Signs and symptoms
It is characterized by a deficiency in biliary copper excretion that causes deformations in the skeleton. These include projections on the back of the skull (parasagittal bone exostoses arising from the occipital bone—the so-called "occipital horns") as well as deformities of the elbow, radial head dislocation, hammer-shaped lateral ends of the clavicles, and abnormalities of the hips and pelvis.[1] OHS presents in early to middle childhood.[4] Children may present with features such as:
- Normal/slightly delayed intelligence
- Long neck, high arched palate, long face, high forehead
- Looseness of skin and "double jointed"
- Inguinal hernias
- Twisting of blood vessels
- Bladder diverticula
- Dysautonomia—inability to regulate parts of the nervous system
- Chronic diarrhea
- Coarse hair
- Low ceruloplasmin levels (9-29mg/dL)[6]
- Damage to the central nervous system [7]
- Muscle wasting[7]
Causes
OHS is a milder allelic variant of Menkes disease, having a later age of onset and being associated with far less severe central neurodegeneration. The milder nature of OHS is often attributable to ‘leaky’ splice junction mutations that allow 20–30% of ATP7A messenger RNA (mRNA) transcripts to be correctly processed. As in cases of Menkes disease, individuals with OHS manifest connective tissue abnormalities resulting from deficient activity of lysyl oxidase, a copper-requiring enzyme that normally deaminates lysine and hydroxylysine in the first step of collagen crosslink formation. Such individuals also often endure inconvenient dysautonomic signs and symptoms related to a partial deficiency in dopamine-β-hydroxylase (DBH) activity. DBH, another copper-dependent enzyme, normally converts dopamine to norepinephrine, a crucial neurotransmitter in norepinephrinergic neurons. A natural mouse model of OHS, the so-called mottled blotchy model, recapitulates the connective tissue abnormalities, DBH deficiency and mild CNS damage seen in humans.[8]
Diagnosis
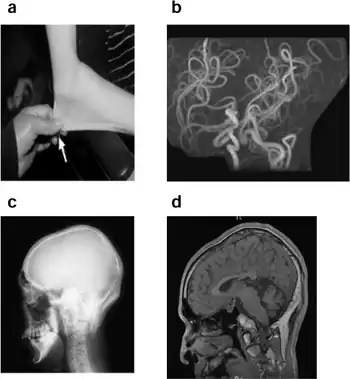
The initial diagnosis of Menkes disease (MD) and its milder variants such as Occipital Horn Syndrome is based on the clinical symptoms. Low serum copper and ceruloplasmin levels support the clinical suspicion of OHS, but biochemical confirmation in tissue culture is needed. The ultimate diagnostic proof is the demonstration of a molecular defect in ATP7A. Demonstration of the bony protuberances on the occiput will clinch the diagnosis, and these can be palpated in some patients.[9]
Treatment
Courses of treatment for children with OHS is dependent upon the severity of their case. Children with OHS often receive physical and occupational therapy.[4] They may require a feeding tube to supplement nourishment if they are not growing enough. In an attempt to improve the neurological condition (seizures) copper histidine or copper chloride injections can be given early in the child's life. However, copper histidine injections have been shown ineffective for treating the connective tissue manifestations of OHS.[10]
Prognosis
The long term natural history of OHS is not known.[8] Some patients have died suddenly as young as 17 years of age,[11] whereas one patient has survived to age 57.[12] Causes of death include respiratory failure,[11] aortic aneurysm,[13] and intracranial hemorrhage.[14]
Research
The NIH and Cyprium Therapeutics are coordinating the joint-development of an Adeno-Associated virus gene therapy named AAV-ATP7A, for Menkes disease and its milder variants such as Occipital Horn Syndrome.[15] In March 2017, Cyprium Therapeutics acquired the World-Wide development and commercial rights to the Menkes program at NIH/NICHD through CRADA and licensing agreements with NICHD.[16] AAV-ATP7A is still in pre-clinical stage, although it has received orphan drug designation from the FDA.
See also
References
- 1 2 Online Mendelian Inheritance in Man (OMIM): 304150
- ↑ Scheiber, Ivo; Dringen, Ralf; Mercer, Julian F. B. (2013). "Chapter 11. Copper: Effects of Deficiency and Overload". In Astrid Sigel, Helmut Sigel and Roland K. O. Sigel (ed.). Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. Vol. 13. Springer. pp. 359–387. doi:10.1007/978-94-007-7500-8_11. ISBN 978-94-007-7499-5. PMID 24470097.
- ↑ Tang J, Robertson S, Lem KE, Godwin SC, Kaler SG (November 2006). "Functional copper transport explains neurologic sparing in occipital horn syndrome". Genetics in Medicine. 8 (11): 711–8. doi:10.1097/01.gim.0000245578.94312.1e. PMID 17108763.
- 1 2 3 Horn Syndrome Archived 2016-10-11 at the Wayback Machine, 9 August 2004.
- ↑ Kennerson ML, Nicholson GA, Kaler SG, Kowalski B, Mercer JF, Tang J, et al. (March 2010). "Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy". American Journal of Human Genetics. 86 (3): 343–52. doi:10.1016/j.ajhg.2010.01.027. PMC 2833394. PMID 20170900.
- ↑ Kodama H, Murata Y, Kobayashi M (August 1999). "Clinical manifestations and treatment of Menkes disease and its variants". Pediatrics International. 41 (4): 423–9. doi:10.1046/j.1442-200x.1999.01095.x. PMID 10453199. S2CID 21267747.
- 1 2 Wakai S, Ishikawa Y, Nagaoka M, Okabe M, Minami R, Hayakawa T (May 1993). "Central nervous system involvement and generalized muscular atrophy in occipital horn syndrome: Ehlers-Danlos type IX. A first Japanese case". Journal of the Neurological Sciences. 116 (1): 1–5. doi:10.1016/0022-510x(93)90081-9. PMID 8099605. S2CID 40538663.
- 1 2 Kaler SG (January 2011). "ATP7A-related copper transport diseases-emerging concepts and future trends". Nature Reviews. Neurology. 7 (1): 15–29. doi:10.1038/nrneurol.2010.180. PMC 4214867. PMID 21221114.
- ↑ Horn N, Tümer Z (2003). "Chapter 14: Menkes Disease and the Occipital Horn Syndrome". In Royce PM, Steinmann B (eds.). Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects (Second ed.). Wiley-Blackwell. pp. 651–685. doi:10.1002/0471221929.ch14. ISBN 978-0-471-25185-9.
- ↑ Kodama H, Fujisawa C, Bhadhprasit W (March 2011). "Pathology, clinical features and treatments of congenital copper metabolic disorders--focus on neurologic aspects". Brain & Development. 33 (3): 243–51. doi:10.1016/j.braindev.2010.10.021. PMID 21112168. S2CID 38032382. Archived from the original on 2022-02-17. Retrieved 2022-01-06.
- 1 2 Yasmeen S, Lund K, De Paepe A, De Bie S, Heiberg A, Silva J, Martins M, Skjørringe T, Møller LB (April 2014). "Occipital horn syndrome and classical Menkes Syndrome caused by deep intronic mutations, leading to the activation of ATP7A pseudo-exon". European Journal of Human Genetics. 22 (4): 517–21. doi:10.1038/ejhg.2013.191. PMC 3953917. PMID 24002164.
- ↑ Bonati MT, Verde F, Hladnik U, Cattelan P, Campana L, Castronovo C, Ticozzi N, Maderna L, Colombrita C, Papa S, Banfi P, Silani V (December 2017). "ATP7A pathogenic variant in a family exhibiting a variable occipital horn syndrome phenotype". Molecular Genetics and Metabolism Reports. 13: 14–17. doi:10.1016/j.ymgmr.2017.07.007. PMC 5522958. PMID 28761814.
- ↑ Quiroga E, Heneghan R (June 2015). "Abdominal aortic aneurysm in a patient with occipital horn syndrome 2". Journal of Vascular Surgery Cases. 1 (2): 138–140. doi:10.1016/j.jvsc.2015.03.012. PMC 6849971. PMID 31725130.
- ↑ Proud VK, Mussell HG, Kaler SG, Young DW, Percy AK (October 1996). "Distinctive Menkes disease variant with occipital horns: delineation of natural history and clinical phenotype". American Journal of Medical Genetics. 65 (1): 44–51. doi:10.1002/(SICI)1096-8628(19961002)65:1<44::AID-AJMG7>3.0.CO;2-Y. PMID 8914740.
- ↑ Donsante A, Yi L, Zerfas PM, Brinster LR, Sullivan P, Goldstein DS, Prohaska J, Centeno JA, Rushing E, Kaler SG (December 2011). "ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model". Molecular Therapy. 19 (12): 2114–23. doi:10.1038/mt.2011.143. PMC 3242653. PMID 21878905.
- ↑ "Corporate Presentation" (PDF). Cyprium Therapeutics, Inc. October 2017. Archived (PDF) from the original on 2022-02-17. Retrieved 2022-01-06.
External links
| Classification | |
|---|---|
| External resources |
|
- GeneReviews/NCBI/NIH/UW entry on ATP7A-Related Copper Transport Disorders Archived 2010-03-21 at the Wayback Machine
- Occipital horn syndrome at NIH's Office of Rare Diseases