Kallmann syndrome
| Kallmann syndrome | |
|---|---|
| Other names: Kallmann's hereditary anosmia, hypogonadism with anosmia, anosmic hypogonadism[1] | |
| Video explanation | |
| Specialty | Endocrinology, medical genetics |
| Symptoms | Decreased sense of smell, lack of sexual development[2] |
| Complications | Osteoporosis, infertility, sexual dysfunction[2] |
| Usual onset | Present at birth[2] |
| Duration | Life-long[3] |
| Causes | Genetic mutation[4] |
| Risk factors | Family history[2] |
| Diagnostic method | Blood hormone levels, genetic testing[2] |
| Differential diagnosis | Isolated hypogonadotropic hypogonadism[2] |
| Treatment | Hormone replacement therapy, gonadotropins[3] |
| Frequency | 1 in 30,000 (males), 1 in 120,000 (females)[1] |
Kallmann syndrome (KS) is a genetic disorder that results in a decreased sense of smell and a lack of sexual development.[2] People may also have hearing loss, teeth abnormalities, and poorly developed kidneys.[2] Other symptoms may include undescended testicles and a small penis in males.[2] Without treatment complications may include osteoporosis, infertility, and sexual dysfunction.[2]
A variety of genetic mutations can cause KS.[3] Often this occurs spontaneously but some cases may be inherited from a person's parents.[2] Inheritance may be X-linked recessive, autosomal dominant, or autosomal recessive.[2] The underlying mechanism is a failure to produce gonadotropin-releasing hormone, resulting in low testosterone in males and estrogen and progesterone in females.[2][3] It is a form of hypogonadotropic hypogonadism.[2] Diagnosis often occurs when puberty fails to start and is confirmed by testing hormone levels or genetic testing.[2]
Treatment is generally with hormone replacement therapy (HRT).[2] This may include testosterone or estrogen and progesterone.[3] Gonadotropin may be used to support fertility.[3] In and of itself, it does not result in a lowered life expectancy, though associated complications may.[3]
Kallmann syndrome affects about 1 in 30,000 males and 1 in 120,000 females.[1] The link between loss of smell and small testicles was first described in 1856 by Spanish doctor Aureliano Maestre de San Juan.[5][6] It was described more fully in 1944 by Franz Josef Kallmann, a German-American geneticist after who it is named.[7][8]
Signs and symptoms


It is normally difficult to distinguish a case of Kallmann syndrome (KS)/hypogonadotropic hypogonadism (HH) from a straightforward constitutional delay of puberty. However, if puberty has not started by either age 14 (girls) or 15 (boys) years and one or more of the non-reproductive features mentioned below is present, then a referral to reproductive endocrinologist might be advisable.[9][10][11]
The features of KS and other forms of HH can be split into two different categories; "reproductive" and "non-reproductive".[12][5][13][14][15]
Reproductive features
- Failure to start or fully complete puberty.[10]
- Lack of testicle development in men (size < 4 ml, whereas the normal range is between 12 and 25 ml).[10]
- Primary amenorrhoea (failure to start menstruation).[11]
- Poorly defined secondary sexual characteristics.[15]
- Micropenis in 5-10% of male cases.[10]
- Cryptorchidism (undescended testicles) at birth.[10]
- Low levels of the gonadotropins LH and FSH.[15]
- Hypogonadism due to low levels of testosterone in men or oestrogen/progesterone in women.[15]
- Infertility.[10]
Non-reproductive features
- Total lack of sense of smell (anosmia) or markedly reduced sense of smell (hyposmia). This is the defining feature of Kallmann syndrome; it is not seen in other cases of HH. Approximately 50% of HH cases occur with anosmia and can be termed as Kallmann syndrome.[15]
- Cleft palate, cleft lip or other midline cranio-facial defects.[12]
- Neural hearing impairment[15]
- Absence of one of the kidneys (unilateral renal agenesis)[15]
- Skeletal defects including split hand/foot (ectrodactyly), shortened middle finger (metacarpal)[15] or scoliosis[16]
- Manual synkinesis (mirror movements of hands)[15]
- Missing teeth (hypodontia)[15]
- Poor balance or coordination due to cerebral ataxia.[11]
- Eye defects such as coloboma or ptosis.[5]
- Increased incidence of color-blindness [17][18]
The exact genetic nature of each particular case of KS/HH will determine which, if any, of the non-reproductive features will occur. The severity of the symptoms will also vary from case to case. Even family members will not show the same range or severity of symptoms.[15][11]
KS/HH is most often present from birth but adult onset versions are found in both males and females. The hypothalamic-pituitary-gonadal axis (HPG axis) functions normally at birth and well into adult life, giving normal puberty and normal reproductive function. The HPG axis then either fails totally or is reduced to a very low level of GnRH release in adult life with no obvious cause (e.g. a pituitary tumour). This will lead to a fall in testosterone or oestrogen levels and infertility.[16][19]
Functional hypothalamic amenorrhoea is seen in females where the HPG axis is suppressed in response to physical or psychological stress or malnutrition but is reversible with the removal of the stressor.[10]
Some cases of KS/HH appear to reverse during adult life where the HPG axis resumes its normal function and GnRH, LH, and FSH levels return to normal levels. This occurs in an estimated 10 to 22% of people, primarily normosmic CHH cases rather than KS cases and only found in people who have undergone some form of testosterone replacement therapy. It is only normally discovered when testicular volume increases while on testosterone treatment alone and testosterone levels return to normal when treatment is stopped. This type of KS/HH rarely occurs in cases where males have had a history of un-descended testes.[11][12]
Affected individuals with KS and other forms of HH are almost invariably born with normal sexual differentiation; i.e., they are physically male or female. This is due to the human chorionic gonadotrophin (hCG) produced by placenta at approximately 12 to 20 weeks gestation (pregnancy) which is normally unaffected by having KS or CHH.[20]
People with KS/HH lack the surge of GnRH, LH, and FSH that normally occurs between birth and six months of age. This surge is particularly important in infant boys as it helps with testicular descent into the scrotum. The surge of GnRH/LH/FSH in non KS/HH children gives detectable levels of testosterone in boys and oestrogen and progesterone in girls. The lack of this surge can sometimes be used as a diagnostic tool if KS/HH is suspected in a newborn boy, but is not normally distinct enough for diagnosis in girls.[12]
Osteoporosis
One possible side effect of having KS/CHH is the increased risk of developing secondary osteoporosis or osteopenia. Oestrogen (females) or testosterone (males) is essential for maintaining bone density.[21] Deficiency in either testosterone or oestrogen can increase the rate of bone resorption while at the same time slowing down the rate of bone formation. Overall this can lead to weakened, fragile bones which have a higher tendency to fracture.
Even a short time with low oestrogen or testosterone, as in cases of delayed diagnosis of KS/CHH can lead to an increased risk of developing osteoporosis but other risk factors, such as smoking are involved so the risk of developing it will vary from person to person. Bone density scans are recommended to monitor the bone mineral density.[16]
The bone density scan is known as a dual energy X-ray absorptiometry scan (DEXA or DXA scan). It is a simple test, taking less than 15 minutes to perform. It involves taking a specialised X-ray picture of the spine and hips and measuring the bone mineral density and comparing the result to the average value for a young healthy adult in the general population.[22]
Adequate calcium levels and, probably, more importantly, vitamin D levels are essential for healthy bone density. Some people with KS/CHH will have their levels checked and may be prescribed extra vitamin D tablets or injections to try to prevent the condition getting worse. The role of vitamin D for general overall health is under close scrutiny at the moment with some researchers claiming vitamin D deficiency is prevalent in many populations and can be linked to other diseases.[23]
Some people with severe osteoporosis might be prescribed bisphosphonates to preserve bone mass, in addition to hormone replacement therapy.[24]
Genetics
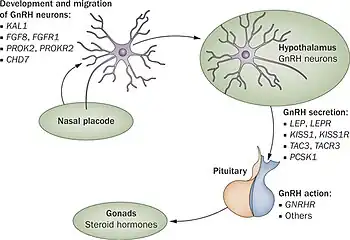
To date at least 25 different genes have been implicated in causing Kallmann syndrome or other forms of hypogonadotropic hypogonadism through a disruption in the production or activity of GnRH (37). These genes involved cover all forms of inheritance and no one gene defect has been shown to be common to all cases which makes genetic testing and inheritance prediction difficult.[25][26]
The number of genes known to cause cases of KS/CHH is still increasing.[14] In addition it is thought that some cases of KS/CHH are caused by two separate gene defects occurring at the same time.[11]
Individual gene defects can be associated with specific symptoms which can help in identifying which genes to test for.[11][15] Between 35-45% of cases of KS/CHH have an unknown genetic cause.[27]
The ANOS1 gene defect (previously known as KAL-1) was the first one discovered and the one most commonly tested for. It causes the x-linked form of Kallmann syndrome and is associated with the additional symptoms of anosmia, bimanual synkinesis and renal agenesis. This defect is thought to be responsible for between 5 and 10% of all Kallmann syndrome/CHH cases.[11][15]
Pathophysiology
The underlying cause of Kallmann syndrome or other forms of hypogonadotropic hypogonadism is a failure in the correct action of the hypothalamic hormone GnRH. The term isolated GnRH deficiency (IGD) has increasingly been used to describe this group of conditions as it highlights the primary cause of these conditions and distinguishes them from other conditions such as Klinefelter syndrome or Turner syndrome which share some similar symptoms but have a different etiology.[28] The term hypogonadism describes a low level of circulating sex hormones; testosterone in males and oestrogen and progesterone in females. Hypogonadism can occur through a number of different mechanisms. The use of the term hypogonadotropic relates to the fact that the hypogonadism found in HH is caused by a disruption in the production of the gonadotropin hormones normally released by the anterior pituitary gland known as luteinising hormone (LH) and follicle stimulating hormone (FSH).[14][27] Failure in GnRH activity can otherwise be due to the absence of the GnRH releasing neurons inside the hypothalamus. HH can occur as an isolated condition with just the LH and FSH production being affected or it can occur in combined pituitary deficiency conditions.
In the first 10 weeks of normal embryonic development, the GnRH releasing neurons migrate from their original source in the nasal region and end up inside the hypothalamus. These neurons originate in an area of the developing head, the olfactory placode, that will give rise to the olfactory epithelium; they then pass through the cribriform plate, along with the fibres of the olfactory nerves, and into the rostral forebrain. From there they migrate to what will become the hypothalamus. Any problems with the development of the olfactory nerve fibres will prevent the progression of the GnRH releasing neurons towards the brain.[29]
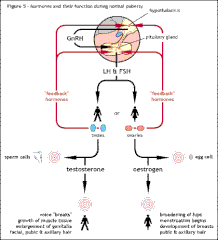 Shows the normal hormonal control of puberty from the hypothalamus down to the testes or ovaries and their negative feedback mechanisms. The negative feedback control allows just the right amount of hormone to be released according to the needs of the body at that time.
Shows the normal hormonal control of puberty from the hypothalamus down to the testes or ovaries and their negative feedback mechanisms. The negative feedback control allows just the right amount of hormone to be released according to the needs of the body at that time.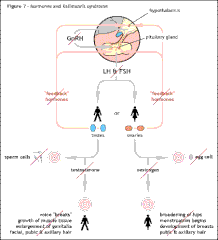 Shows the effect of the interruption of GnRH hormone release from the hypothalamus and the subsequent inability of the testes and ovaries to function correctly at puberty as seen in cases of KS/HH. In most cases of KS/HH the testes and ovaries are able to function correctly, but fail to do so because they have not had the correct hormonal signals.
Shows the effect of the interruption of GnRH hormone release from the hypothalamus and the subsequent inability of the testes and ovaries to function correctly at puberty as seen in cases of KS/HH. In most cases of KS/HH the testes and ovaries are able to function correctly, but fail to do so because they have not had the correct hormonal signals.
Diagnosis

Diagnosing KS and other forms of CHH is complicated by the difficulties in distinguishing between a normal constitutional delay of puberty or a case of KS/CHH.[30][13][31] The diagnosis is often one of exclusion found during the workup of delayed puberty.[32][33][34]
In males, the use of age appropriate levels of testosterone can help to distinguish between a case of KS/CHH from a case of delayed puberty. If no puberty is apparent, especially no testicular development, then a review by a reproductive endocrinologist may be appropriate. If puberty is not apparent by the age of 16 then the person should be referred for endocrinological review.[35] Post natal diagnosis of KS/CHH before the age of 6 months is sometimes possible as the normal post natal hormonal surge of gonadotropins along with testosterone or oestrogen is absent in babies with KS/CHH. This lack of detectable hormones in the blood can be used as a diagnostic indicator, especially in male infants.[36]
In females, diagnosis is sometimes further delayed as other causes of amenorrhoea normally have to be investigated first before a case of KS/CHH is considered.[37]
Diagnosis of KS/CHH normal involves a range of clinical, biochemical and radiological tests to exclude other conditions that can cause similar symptoms.
Clinical tests
- Comparing height to standard growth charts.
- Determining the Tanner stage of sexual development. (Males with KS/CHH are normally at stage I or II with genitalia, females at stage I with breast development and both males and females at stage III with pubic hair development).[15]
- Checking for micropenis and undescended testes (cryptorchidism) in males.
- Measuring testicular volume.
- Checking for breast development and age at menarche in females.
- Checking sense of smell using odorant panel or University of Pennsylvania Smell Identification Test (UPSIT)
- Checking for hearing impairment.
- Checking for missing teeth or presence of cleft lip and/or cleft palate.
- Checking for pigmentation of skin and hair.
- Checking for mirror movements of the hands or signs of neurodevelopmental delay.
Lab tests
- Early morning hormonal testing including FSH, LH, testosterone, oestrogen and prolactin.
- GnRH and/or hCG stimulation test to determine activity of hypothalamus and pituitary.
- Sperm test
- Liver function, renal function and inflammation marker testing.
- Karyotype to check for chromosomal abnormalities.
Medical imaging
- Performing wrist x-ray to determine bone age.
- Brain MRI to rule out any structural abnormalities in the hypothalamus or pituitary and to check for presence of olfactory bulbs.
- Ultrasound of kidneys to rule out unilateral renal agenesis.
- Bone density scan (DXA) to check for osteoporosis or osteopenia.
Treatment
For both males and females, the initial aim for treatment is the development of the secondary sexual characteristics normally seen at puberty.[15][38][33][34][39] Once this has been achieved, continued hormone replacement therapy is required for both males and females to maintain sexual function, bone health, libido and general wellbeing.[12] In males, testosterone replacement therapy is required for the maintenance of normal muscle mass.[15]
Early treatment is sometimes required for male infants with suspected KS/CHH to correct un-descended testes and micropenis if present with the use or surgery or gonadotropin or DHT treatment. Females with KS/CHH normally do not require any treatment before the age of adolescence. Currently, no treatments exist for the lack of sense of smell, mirror movement of the hands or the absence of one kidney.[12]
Treatment for both males and females with KS/CHH normally consists of one of three options which can be used for both hormone replacement therapy and/or fertility treatment.[15][12]
- Sex hormone replacement (testosterone or oestrogen & progesterone).
- Gonadotropin therapy (medications that replicate the activity of FSH and LH).
- GnRH pulsatile therapy.
Hormone replacement therapy
The method and dose of treatment will vary depending on the individual being treated. Initial treatment is normally made with lower doses in younger patients in order to develop the secondary sexual characteristics before adult doses are reached.[15]
For males with KS/CHH the types of testosterone delivery include daily patches, daily gel use, daily capsules, sub cutaneous or intramuscular injections or six-monthly implants. Different formulations of testosterone are used to ensure both the anabolic and androgenic effects of testosterone are achieved.[12][13] Nasal testosterone delivery methods have been developed but their use in KS/CHH treatment has not been formally evaluated.[15]
Gonadotropin therapy, in the form of human chorionic gonadotropin (hCG) injections, with or without the use of FSH, can also be used in male patients to induce secondary sexual characteristic development alongside possible fertility induction.[12]
For females, hormone replacement involves the use of oestrogen and progesterone. Firstly, oestrogen is used in tablet or gel form in order to maximise breast development, then a combination of oestrogen and progesterone is used.[12][15] Cyclical progesterone is normally required to help keep the endometrium (lining of the uterus) healthy.[15]
In males, the monitoring of treatment normally requires the measurement of serum testosterone, inhibin B, haematocrit and prostate-specific antigen (PSA). If injections are used, trough levels are taken to ensure an adequate level of testosterone is achieved throughout the injection cycle.[12]
In females monitoring normally consists of measurement of oestrogen, FSH, LH, inhibin B and anti-Müllerian hormone (AMH).[12]
Standard hormone replacement therapy will not normally induce fertility in either males or females, with no testicular growth in males. Early treatment as adolescents can help with psychological well being of people with KS/CHH.[12]
Fertility treatments
Gonadotropin therapy can be used in both male and female patients in order to achieve fertility for some people.[12][15]
Pulsatile GnRH therapy can also be used to induce fertility, especially in females, but its use is limited to a few specialist treatment centres.[15]
In males with KS/CHH, infertility is primarily due to the lack of sperm production within the testes. Sperm production can be achieved through either the use of GnRH administered via a micro infusion pump or through the use of gonadotropin injections (hCG, FSH, hMG). The time taken to achieve adequate sperm production for natural conception will vary from person to person. If the pre-treatment testes are very small and there has been a history of undescended testes it might take longer to achieve sperm production. In these cases, assisted reproductive technology, such as sperm retrieval using testicular sperm extraction (TESE) and/or intracytoplasmic sperm injection (ICSI), might be required.[40]
In females with KS/CHH, infertility is primarily due to the lack of maturation of eggs located within the ovaries. Ovulation induction can be achieved either with pulsatile GnRH therapy or alternatively with gonadotropin injections (hCG, FSH, hMG) given at set intervals to trigger the maturation and release of the egg for natural conception.[40]
Prognosis
Reversal of symptoms has been reported in between 10% to 22% of cases.[41][15]
Reversal cases have been seen in both KS and normosmic CHH but appear to be less common in cases of KS (where the sense of smell is also affected). Reversal is not always permanent and the precise genetic causes are not yet fully understood.[42]
Epidemiology
The epidemiology of Kallmann syndrome is not well understood. Individual studies include a 1986 report reviewing medical records in the Sardinian army which found a prevalence of 1 in 86,000 men[43] and a 2011 report from Finland which found a prevalence of 1:30,000 for males and 1:125,000 for females.[44]
Kallmann syndrome occurs about 4 times more often in males than females, but is only 2.5 times more common among males in familial cases.[43][44]
History

Kallmann syndrome was first described by name in a paper published in 1944 by Franz Josef Kallmann, a German-American geneticist.[8][45] The link between anosmia and hypogonadism had already been noted by the Spanish doctor Aureliano Maestre de San Juan in 1856.[6] In the 1950s, De Morsier and Gauthier reported the partial or complete absence of the olfactory bulb in the brains of men with hypogonadism.[46][5]
Terminology
The terminology used when describing cases of HH vary and can include:
- GnRH deficiency
- congenital hypogonadotropic hypogonadism (CHH)[47]
- idiopathic/isolated hypogonadotropic hypogonadism (IHH)
- normosmic hypogonadotropic hypogonadism (nHH)
- hypothalamic hypogonadism
- olfacto-genital syndrome
Research
Kisspeptin is a protein that regulates the release of GnRH from the hypothalamus, which in turn regulates the release of LH and, to a lesser extent, FSH from the anterior pituitary gland. Kisspeptin and its associated receptor KISS1R are known to be involved in the regulation of puberty. Studies have shown there is potential for kisspeptin to be used in the diagnosis and treatment of certain cases of Kallmann syndrome and CHH.[48][49]
References
- 1 2 3 "Kallmann syndrome: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 5 March 2021. Retrieved 22 January 2021.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 "Kallmann syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 13 February 2021. Retrieved 21 January 2021.
- 1 2 3 4 5 6 7 Sonne, J; Lopez-Ojeda, W (January 2020). "Kallmann Syndrome". PMID 30855798.
{{cite journal}}: Cite journal requires|journal=(help) - ↑
- 1 2 3 4 Kim SH (December 2015). "Congenital Hypogonadotropic Hypogonadism and Kallmann Syndrome: Past, Present, and Future". Endocrinology and Metabolism. 30 (4): 456–66. doi:10.3803/EnM.2015.30.4.456. PMC 4722398. PMID 26790381.
- 1 2 Maestre de San Juan, Aureliano (1856). "Teratolagia: falta total de los nervios olfactorios con anosmia en un individuo en quien existia una atrofia congenita de los testiculos y miembro viril". El Siglo Médico. 3: 211–221.
- ↑ "Kallmann's syndrome". www.whonamedit.com. Archived from the original on 3 May 2019. Retrieved 22 January 2021.
- 1 2 Kallmann FJ, Schönfeld WA, Barrera SE (1943–1944). "The genetic aspects of primary eunuchoidism". Am J Ment Defic. 48: 203–236.
- ↑ McCabe MJ, Bancalari RE, Dattani MT (February 2014). "Diagnosis and evaluation of hypogonadism". Pediatric Endocrinology Reviews. 11 Suppl 2 (Feb): 214–29. PMID 24683946.
- 1 2 3 4 5 6 7 "Kallmann syndrome". Genetics Home Reference. US Library of Medicine. National Institutes for Health. Genetic and Rare Diseases Information. June 26, 2016. Archived from the original on December 16, 2017. Retrieved December 17, 2017.
- 1 2 3 4 5 6 7 8 Lima Amato LG, Latronico AC, Gontijo Silveira LF (June 2017). "Molecular and Genetic Aspects of Congenital Isolated Hypogonadotropic Hypogonadism". Endocrinology and Metabolism Clinics of North America. 46 (2): 283–303. doi:10.1016/j.ecl.2017.01.010. PMID 28476224.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J (September 2015). "Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment". Nature Reviews. Endocrinology. 11 (9): 547–64. doi:10.1038/nrendo.2015.112. PMID 26194704.
- 1 2 3 Dunkel L, Quinton R (June 2014). "Transition in endocrinology: induction of puberty". European Journal of Endocrinology. 170 (6): R229–39. doi:10.1530/EJE-13-0894. PMID 24836550.
- 1 2 3 Mitchell AL, Dwyer A, Pitteloud N, Quinton R (July 2011). "Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory". Trends in Endocrinology and Metabolism. 22 (7): 249–58. doi:10.1016/j.tem.2011.03.002. PMID 21511493. S2CID 23578201.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 Balasubramanian R, Crowley WF Jr (2017). "Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency". SourceGeneReviews. PMID 20301509.
- 1 2 3 "Kallmann syndrome". Rare Diseases. National Organisation for Rare Disorders (NORD). 2012. Archived from the original on December 27, 2017. Retrieved December 16, 2017.
- ↑ Chopra R, Chander A, Jacob JJ (May 2012). "The eye as a window to rare endocrine disorders". Indian Journal of Endocrinology and Metabolism. 16 (3): 331–8. doi:10.4103/2230-8210.95659. PMC 3354836. PMID 22629495.
- ↑ Jaffe MJ, Sherins RJ, de Monasterio F (1989). Colour Vision Deficiencies IX. Documenta Ophthalmologica Proceedings Series. Dordrecht: Springer. pp. 201–207. doi:10.1007/978-94-009-2695-0_24. ISBN 9789401077156.
- ↑ "Kallmann syndrome". National Institutes for Health. US Library of Medicine. Genetics Home Reference. December 2017. Archived from the original on December 16, 2017. Retrieved December 17, 2017.
- ↑ Sperling, Mark (2014). Pediatric Endocrinology E-Book. Elsevier Health Sciences. p. 136. ISBN 9781455759736.
- ↑ Guo CY, Jones TH, Eastell R (February 1997). "Treatment of isolated hypogonadotropic hypogonadism effect on bone mineral density and bone turnover". The Journal of Clinical Endocrinology and Metabolism. 82 (2): 658–65. doi:10.1210/jc.82.2.658. PMID 9024272.
- ↑ Laitinen EM, Hero M, Vaaralahti K, Tommiska J, Raivio T (August 2012). "Bone mineral density, body composition and bone turnover in patients with congenital hypogonadotropic hypogonadism". International Journal of Andrology. 35 (4): 534–40. doi:10.1111/j.1365-2605.2011.01237.x. PMID 22248317.
- ↑ Wimalawansa SJ, Razzaque DM, Al-Daghri NM (December 2017). "Calcium and Vitamin D in Human Health: Hype or Real?". The Journal of Steroid Biochemistry and Molecular Biology. Dec 16: 4–14. doi:10.1016/j.jsbmb.2017.12.009. PMID 29258769. S2CID 11467429.
- ↑ Golds G, Houdek D, Arnason T (2017). "Male Hypogonadism and Osteoporosis: The Effects, Clinical Consequences, and Treatment of Testosterone Deficiency in Bone Health". Int J Endocrinol. 2017: 4602129. doi:10.1155/2017/4602129. PMC 5376477. PMID 28408926.
- ↑ Layman LC (May 2013). "Clinical genetic testing for Kallmann syndrome". The Journal of Clinical Endocrinology and Metabolism. 98 (5): 1860–2. doi:10.1210/jc.2013-1624. PMC 3644595. PMID 23650337.
- ↑ Valdes-Socin H, Rubio Almanza M, Tomé Fernández-Ladreda M, Debray FG, Bours V, Beckers A (2014). "Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes". Frontiers in Endocrinology. 5 (109): 109. doi:10.3389/fendo.2014.00109. PMC 4088923. PMID 25071724.
- 1 2 Vezzoli V, Duminuco P, Bassi I, Guizzardi F, Persani L, Bonomi M (June 2016). "The complex genetic basis of congenital hypogonadotropic hypogonadism". Minerva Endocrinologica. 41 (2): 223–39. PMID 26934720.
- ↑ Au MG, Crowley WF, Buck CL (October 2011). "Genetic counseling for isolated GnRH deficiency". Molecular and Cellular Endocrinology. 346 (1–2): 102–9. doi:10.1016/j.mce.2011.05.041. PMC 3185214. PMID 21664415.
- ↑ Teixeira L, Guimiot F, Dodé C, Fallet-Bianco C, Millar RP, Delezoide AL, Hardelin JP (October 2010). "Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions". The Journal of Clinical Investigation. 120 (10): 3668–72. doi:10.1172/JCI43699. PMC 2947242. PMID 20940512.
- ↑ Pitteloud N (December 2012). "Managing delayed or altered puberty in boys". BMJ. 345 (Dec 3): e7913. doi:10.1136/bmj.e7913. PMID 23207503. S2CID 5159169.
- ↑ Young J (March 2012). "Approach to the male patient with congenital hypogonadotropic hypogonadism". The Journal of Clinical Endocrinology and Metabolism. 97 (3): 707–18. doi:10.1210/jc.2011-1664. PMID 22392951.
- ↑ Lee PA, Houk CP (August 13, 2012). "The Smallest Kid in School: Evaluating Delayed Puberty". Medscape Pediatrics. Archived from the original on January 11, 2015. Retrieved October 4, 2015.
- 1 2 Jones H, ed. (2008). "Chapter 9: Puberty & Fertility". Testosterone Deficiency in Men. Oxford Endocrinology Library. ISBN 978-0199545131.
- 1 2 Jockenhovel F (2004). "Chapter 3: Diagnostic work up of hypogonadism". Male Hypogonadism. Uni-Med Science. ISBN 978-3-89599-748-8.
- ↑ Quinton R (April 2005). "Adolescent development: advice in ABC of adolescence is potentially misleading". BMJ. 330 (7494): 789, author reply 789. doi:10.1136/bmj.330.7494.789. PMC 555895. PMID 15802728.
- ↑ Dwyer AA, Jayasena CN, Quinton R (June 2016). "Congenital hypogonadotropic hypogonadism: implications of absent mini-puberty". Minerva Endocrinologica. 41 (2): 188–95. PMID 27213784.
- ↑ Bry-Gauillard H, Trabado S, Bouligand J, Sarfati J, Francou B, Salenave S, Chanson P, Brailly-Tabard S, Guiochon-Mantel A, Young J (May 2010). "Congenital hypogonadotropic hypogonadism in females: clinical spectrum, evaluation and genetics". Annales d'Endocrinologie. 71 (3): 158–62. doi:10.1016/j.ando.2010.02.024. PMID 20363464.
- ↑ Bouvattier C, Maione L, Bouligand J, Dodé C, Guiochon-Mantel A, Young J (October 2011). "Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism". Nature Reviews. Endocrinology. 8 (3): 172–82. doi:10.1038/nrendo.2011.164. PMID 22009162. S2CID 4564169.
- ↑ Han TS, Bouloux PM (June 2010). "What is the optimal therapy for young males with hypogonadotropic hypogonadism?". Clinical Endocrinology. 72 (6): 731–7. doi:10.1111/j.1365-2265.2009.03746.x. PMID 19912242.
- 1 2 Maione L, Dwyer AA, Francou B, Guiochon-Mantel A, Binart N, Bouligand J, Young J (March 2018). "GENETICS IN ENDOCRINOLOGY: Genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing". European Journal of Endocrinology. 178 (3): R55–R80. doi:10.1530/EJE-17-0749. PMID 29330225.
- ↑ Sidhoum VF, Chan YM, Lippincott MF, Balasubramanian R, Quinton R, Plummer L, Dwyer A, Pitteloud N, Hayes FJ, Hall JE, Martin KA, Boepple PA, Seminara SB (March 2014). "Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system". The Journal of Clinical Endocrinology and Metabolism. 99 (3): 861–70. doi:10.1210/jc.2013-2809. PMC 3942233. PMID 24423288.
- ↑ Dwyer AA, Raivio T, Pitteloud N (June 2016). "MANAGEMENT OF ENDOCRINE DISEASE: Reversible hypogonadotropic hypogonadism". European Journal of Endocrinology. 174 (6): R267–74. doi:10.1530/EJE-15-1033. PMID 26792935.
- 1 2 Tritos, Nicholas A (October 10, 2016). "Kallmann Syndrome and Idiopathic Hypogonadotropic Hypogonadism: Background, Pathophysiology, Epidemiology". eMedicine. Archived from the original on August 29, 2020. Retrieved November 7, 2017.
{{cite journal}}: Cite journal requires|journal=(help) - 1 2 Balasubramanian R, Crowley WF (March 2, 2017). "Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency". GeneReviews. University of Washington, Seattle. PMID 20301509. Archived from the original on November 19, 2017. Retrieved November 7, 2017.
- ↑
- ↑ De Morsier G, Gauthier G (November 1963). "[Olfacto-Genital Dysplasia]". Pathologie et Biologie. 11: 1267–72. PMID 14099201.
- ↑ Valdes-Socin H, Rubio Almanza M, Tomé Fernández-Ladreda M, Debray FG, Bours V, Beckers A (2014). "Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes". Frontiers in Endocrinology. 5: 109. doi:10.3389/fendo.2014.00109. PMC 4088923. PMID 25071724.
- ↑ Skorupskaite K, George JT, Anderson RA (2014). "The kisspeptin-GnRH pathway in human reproductive health and disease". Human Reproduction Update. 20 (4): 485–500. doi:10.1093/humupd/dmu009. PMC 4063702. PMID 24615662.
- ↑ George JT, Seminara SB (November 2012). "Kisspeptin and the hypothalamic control of reproduction: lessons from the human". Endocrinology. 153 (11): 5130–6. doi:10.1210/en.2012-1429. PMC 3473216. PMID 23015291.
External links
- National Organisation for Rare Diseases page on Kallmann syndrome. Archived 2017-12-27 at the Wayback Machine
| Classification | |
|---|---|
| External resources |