Leydig cell hypoplasia
| Leydig cell hypoplasia | |
|---|---|
| Other names | 46,XY DSD due to luteinizing hormone resistance or luteinizing hormone beta subunit deficiency |
 | |
| This condition is inherited in an autosomal recessive manner[1] | |
Leydig cell hypoplasia (or aplasia) (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 genetic males. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism (partially or fully underdeveloped genitalia), hypergonadotropic hypogonadism (decreased or lack of production of sex steroids by the gonads despite high circulating levels of gonadotropins), reduced or absent puberty (lack of development of secondary sexual characteristics, resulting in sexual infantilism if left untreated), and infertility.[2][3]
Leydig cell hypoplasia does not occur in biological females as they do not have either Leydig cells or testicles. However, the cause of the condition in males, luteinizing hormone insensitivity, does affect females, and because LH plays a role in the female reproductive system, it can result in primary amenorrhea or oligomenorrhea (absent or reduced menstruation), infertility due to anovulation, and ovarian cysts.[2][4]
A related condition is follicle-stimulating hormone (FSH) insensitivity, which presents with similar symptoms to those of Leydig cell hypoplasia but with the symptoms in the respective sexes reversed (i.e., hypogonadism and sexual infantilism in females and merely problems with fertility in males). Despite their similar causes, FSH insensitivity is considerably less common in comparison to LH insensitivity.[5]
Symptoms and signs
The symptoms of Leydig cell hypoplasia include pseudohermaphroditism, i.e., feminized, ambiguous, or relatively mildly underdeveloped (e.g., micropenis, severe hypospadias,[6] and/or cryptorchidism [undescended testes]) external genitalia, a female gender identity or gender variance, hypergonadotropic hypogonadism (hypogonadism despite high levels of gonadotropins), delayed, impaired, or fully absent puberty with an associated reduction in or complete lack of development of secondary sexual characteristics (sexual infantilism), impaired fertility or complete sterility, tall stature (due to delayed epiphyseal closure), eunuchoid skeletal proportions, delayed or absent bone maturation, and osteoporosis.[2][3]
Cause
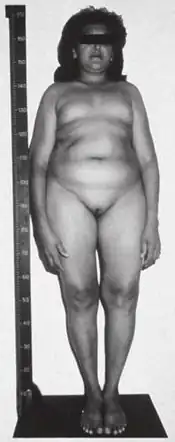
Leydig cell hypoplasia is caused by genetic mutations in LHCGR, a gene which encodes the LH/hCG receptor. LH normally acts through the LH/hCG receptor to stimulate the growth of Leydig cells in the testicles and the production of androgens such as testosterone and dihydrotestosterone (DHT) by these cells. In Leydig cell hypoplasia however, there is a reduced capacity for the LH/hCG receptor to respond to LH. This results in hypoplasia or absence of Leydig cells, testicular atrophy, and lower than normal androgen levels. In the most severe form of the condition in which there is a complete lack of response of the Leydig cells to LH, androgen production by the testicles is virtually negligible and secondary sexual characteristics entirely fail to develop at puberty.[2][3][7][8][9]
Diagnosis
Since the Sertoli cells are not affected by Leydig cell hypoplasia, anti-Müllerian hormone is secreted normally and so there are no Müllerian structures. Wolffian structures, such as the prostate, vasa deferentia, and epidydimides are present. In type I, abdominal testes are revealed on ultrasound; in type II testes may be descended or undescended.
People with Leydig cell hypoplasia type I display no response to the hCG stimulation test; there is no increase in serum levels of testosterone and dihydrotestosterone.[10] Leydig cell hypoplasia type II can display either a pronounced rise of testosterone levels or no rise.
In any case, the diagnosis is confirmed on biopsy of the testes, revealing either absent or hypoplastic Leydig cells. The inside of the testis will be grayish and mucous, displaying arrested spermatogenesis and the presence of Sertoli cells.[11] The diagnosis can also be confirmed by looking for mutations in the gene for the LH receptor.[12]
A diagnosis of Leydig cell hypoplasia is usually made in the neonatal period, following the discovery of ambiguous genitalia, or at puberty, when secondary sex characteristics fail to develop. Puberty is the most common time for Leydig cell hypoplasia to be diagnosed.[11][13]
Treatment
Patients with Leydig cell hypoplasia may be treated with hormone replacement therapy (i.e., with androgens), which will result in normal sexual development and the resolution of most symptoms. In the case of 46,XY (genetically "male") individuals who are phenotypically female and/or identify as the female gender, estrogens should be given instead. Surgical correction of the genitals in 46,XY males may be required, and, if necessary, an orchidopexy (relocation of the undescended testes to the scrotum) may be performed as well.[3]
See also
- Disorders of sex development
- Intersexuality, pseudohermaphroditism, and ambiguous genitalia
- Hypogonadism and hypogonadotropic hypogonadism
- Familial male-limited precocious puberty (or LH oversensitivity)
- Follicle-stimulating hormone insensitivity
- Gonadotropin-releasing hormone insensitivity
- Inborn errors of steroid metabolism
- Isolated 17,20-lyase deficiency
- Combined 17α-hydroxylase/17,20-lyase deficiency
- 17β-Hydroxysteroid dehydrogenase III deficiency
- Androgen insensitivity syndrome
References
- ↑ "OMIM Entry - # 238320 - LEYDIG CELL HYPOPLASIA, TYPE I". omim.org. Retrieved 18 August 2017.
- 1 2 3 4 Wu SM, Leschek EW, Rennert OM, Chan WY (March 2000). "Luteinizing hormone receptor mutations in disorders of sexual development and cancer". Frontiers in Bioscience. 5: D343–52. doi:10.2741/wu. PMID 10704433.
- 1 2 3 4 Eberhard Nieschlag; Hermann M. Behre; Susan Nieschlag (3 December 2009). Andrology: Male Reproductive Health and Dysfunction. Springer. p. 224. ISBN 978-3-540-78354-1. Retrieved 5 June 2012.
- ↑ Arnhold IJ, Latronico AC, Batista MC, Mendonca BB (April 1999). "Menstrual disorders and infertility caused by inactivating mutations of the luteinizing hormone receptor gene". Fertility and Sterility. 71 (4): 597–601. doi:10.1016/s0015-0282(98)00517-2. PMID 10202864.
- ↑ Mark A. Sperling (25 April 2008). Pediatric Endocrinology E-Book. Elsevier Health Sciences. p. 35. ISBN 978-1-4377-1109-7. Retrieved 10 June 2012.
- ↑ Fima Lifshitz (February 2007). Pediatric Endocrinology. CRC Press. p. 374. ISBN 978-1-4200-5523-8. Retrieved 5 June 2012.
- ↑ Themmen AP, Verhoef-Post M (August 2002). "LH receptor defects". Seminars in Reproductive Medicine. 20 (3): 199–204. doi:10.1055/s-2002-35384. PMID 12428200.
- ↑ Mendonca BB, Costa EM, Belgorosky A, Rivarola MA, Domenice S (April 2010). "46,XY DSD due to impaired androgen production". Best Practice & Research. Clinical Endocrinology & Metabolism. 24 (2): 243–62. doi:10.1016/j.beem.2009.11.003. PMID 20541150.
- ↑ Latronico AC, Arnhold IJ (September 2006). "Inactivating mutations of LH and FSH receptors--from genotype to phenotype". Pediatric Endocrinology Reviews. 4 (1): 28–31. PMID 17021580.
- ↑ Amesse, Lawrence S. & Pfaff-Amesse, Teresa (2007). "Chapter 12: Congenital Anomalies of the Female Reproductive Tract". In Falcone, Tomasso & Hurd, William W. (eds.). Clinical Reproductive Medicine and Surgery. Elsevier Health Sciences. p. 184. ISBN 978-0-323-03309-1.
- 1 2 Nistal, Manuel & González-Peramato, Pilar (2016). "Congenital Lesions". In Colecchia, Maurizio (ed.). Pathology of Testicular and Penile Neoplasms. Springer. p. 184. ISBN 978-3-319-27617-5.
- ↑ Nieschlag, Eberhard & Behre, Hermann (June 29, 2013), "Chapter 8: Disorders at the Testicular Level", Andrology: Male Reproductive Health and Dysfunction, Springer Science & Business Media, p. 166, ISBN 978-3-662-04491-9
- ↑ McCann-Crosby, Bonnie & Sutton, V. Reid (2015). "Disorders of Sexual Development". In Gambello, Michael J. & Sutton, V. Reid (eds.). Genetics Diagnosis, Inborn Errors of Metabolism and Newborn Screening: An Update. Elsevier Health Sciences. p. 407. ISBN 978-0-323-35685-5.