Muenke syndrome
| Muenke syndrome | |
|---|---|
| Other names: FGFR3-related craniosynostosis | |
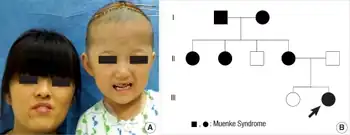 | |
| a) The child and her mother with a Pro250Arg mutation in exon 7 (IgII-IGIII linker domain) of the FGFR3 gene. The mother also had mild midfacial hypoplasia, hypertelorism, and downslanting palpebral fissures. b) pedigree of the family with Muenke syndrome | |
Muenke syndrome, also known as FGFR3-related craniosynostosis,[1] is a human specific condition characterized by the premature closure of certain bones of the skull during development, which affects the shape of the head and face. First described by Maximilian Muenke, the syndrome occurs in about 1 in 30,000 newborns. This condition accounts for an estimated 8 percent of all cases of craniosynostosis.
Signs and symptoms
Many people with this disorder have a premature fusion of skull bones along the coronal suture. Not every case has had craniosynostosis however. Other parts of the skull may be malformed as well. This will usually cause an abnormally shaped head, wide-set eyes, low set ears and flattened cheekbones in these patients. About 5 percent of affected individuals have an enlarged head (macrocephaly). There may also be associated hearing loss in 10–33% of cases and it is important for affected individuals to have hearing tests to check on the possibility of a problem. They can lose about 33-100% of hearing.
Most people with this condition have normal intellect, but developmental delay and learning disabilities are possible. The signs and symptoms of Muenke syndrome vary among affected people, and some findings overlap with those seen in other craniosynostosis syndromes. Between 6 percent and 7 percent of people with the gene mutation associated with Muenke syndrome do not have any of the characteristic features of the disorder.
Other Implications of Muenke Syndrome
Apart from craniosynostosis, it has been suggested that hearing loss, and learning difficulties are common in Muenke syndrome. According to Ulster Medical Journal, most individuals with Muenke syndrome may have limb findings. The most common ocular finding in Muenke syndrome is strabismus as studied by Dr. Agochukwu-Nwubah and her researching team. Approximately 20% of people with Muenke Syndrome will also be affected by epilepsy.
Causes
Muenke syndrome is caused by a specific gene mutation in the FGFR3 gene. The mutation arises randomly; there is no full understanding for what causes this mutation. This mutation causes the FGFR3 protein to be overly active; it interferes with normal bone growth, and allows skull bones to fuse prematurely. There is no connection between anything mother did (or did not do) to activate the syndrome. If neither of the parents have Muenke syndrome, chances of having another child with the syndrome are minimal. This condition is inherited in an autosomal dominant pattern. This means if a parent has Muenke syndrome, every newborn has a 50% chance of inheriting the syndrome.
Genetics

Muenke syndrome is inherited in an autosomal dominant pattern. In some cases, an affected person inherits the mutation from one affected parent. If a patient is shown to have Muenke, they have a 50/50 chance of passing it on to their children. Not all cases of Muenke however is obvious. Other cases may result from new mutations in the gene. These cases occur in people with no history of the disorder in their family.
A single mutation in the FGFR3 gene cause this syndrome. The FGFR3 gene provides instructions for making a protein that is involved in the development and maintenance of bone and brain tissue. This mutation causes the FGFR3 protein to be overly active, which interferes with normal bone growth and allows the bones of the skull to fuse before they should.
As stated by researchers at the University of Washington, Muenke syndrome is inherited in an autosomal dominant manner with incomplete penetrance and variable expressivity.” Prenatal diagnosis for pregnancies at increased risk is possible if the defining mutation has been identified in the family (Agochukwu et.al. 2006). According to the article Craniosynostosis: Molecular Genetics, penetrance is higher in females (87%) than in males (76%). Muenke syndrome is estimated to account for 25%-30% of all genetic causes of craniosynostosis according to the Journal of Anatomy.
Diagnosis
The diagnosis of Muenke syndrome is suspected bases on abnormal skull shape and a diagnosis of coronal craniosynostosis. In 2006, Agochukwu and her colleagues concluded that “A distinct Muenke syndrome phenotype includes: uni or bilateral coronal synostosis, midface hypoplasia, broad toes, and brachydactyly.” Due to phenotypic overlap and/or mild phenotypes, clinical differentiation of this syndrome may be difficult. The suspected diagnosis is confirmed by a blood test to check for gene mutation. To establish the extent of disease in an individual diagnosed with Muenke syndrome, various evaluations are recommended.
Treatment
The treatment of Muenke syndrome is focused on the correction of the abnormal skull shape and mirrors the treatment of coronal craniosynostosis. The abnormal growth patterns continue throughout the growing years; therefore, intervention, accurate diagnosis, and a customized, expertly carried-out treatment plan should be a primary concern. Although the timing of surgery can be highly individualized, surgical correction of the bicoronal craniosynostosis is most often done between 6 and 12 months of age. Surgery is usually performed through a scalp incision that lies concealed within the hair of the head. Your craniofacial surgeon will work in concert with a pediatric neurosurgeon in order to safely remove the bones of the skull. Then, the craniofacial surgeon reshapes and repositions those bones to give a more normal skull shape.
References
- ↑ Clinical trial number NCT00106977 for "Clinical Study of Muenke Syndrome (FGFR3-Related Craniosynostosis)" at ClinicalTrials.gov
- Rannan-Eliya SV, Taylor IB, De Heer IM, Van Den Ouweland AM, Wall SA, Wilkie AO (August 2004). "Paternal origin of FGFR3 mutations in Muenke-type craniosynostosis". Hum. Genet. 115 (3): 200–7. doi:10.1007/s00439-004-1151-5. PMID 15241680. S2CID 24791706.
- Agochukwu NB, Doherty ES, Muenke M (December 7, 2010). "Muenke Syndrome". Gene Reviews. PMID 20301588. Archived from the original on March 3, 2022. Retrieved May 17, 2022.
- Hughes J, Nevin NC, Morrison PJ (2001). "Familial craniosynostosis due to Pro250Arg mutation in the fibroblast growth factor receptor 3 gene". Ulster Med J. 70 (70): 47–50. PMC 2449219. PMID 11428324.
- Morris-Kay GM, Wilkie AO (2005). "Growth of the normal skull vault and its alteration in craniosynostosis: Insights from human genetics and experimental studies". Journal of Anatomy. 207 (5): 637–653. doi:10.1111/j.1469-7580.2005.00475.x. PMC 1571561. PMID 16313397.
- Solomon BD, Muenke M (2010). "Craniosynostosis: Molecular Genetics". Principles of Diagnosis and Treatment. 19: 89–97.
External links
- National Institute of Health's Reference Archived 2010-04-08 at the Wayback Machine
- GeneReview/NIH/UW entry on Muenke Syndrome Archived 2022-03-03 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|