Junctional epidermolysis bullosa (medicine)
| Junctional epidermolysis bullosa (medicine) | |
|---|---|
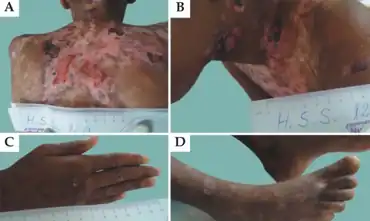 | |
| a-d)Lesions in individuals with junctional epidermolysis bullosa | |
| Specialty | Dermatology |
Junctional epidermolysis bullosa is a skin condition characterized by blister formation within the lamina lucida of the basement membrane zone.[1]: 599 [2]
Signs and symptoms
Sufferers experience very fragile skin, with blisters and skin erosion occurring in response to relatively benign trauma. Blisters may form all over the body, including the mucous membranes. Chronic scarring can lead to the formation of granulation tissue, which may bleed easily, predisposing to infection. Hands and fingers may be affected, as well as various joints.[3]
Pathophysiology
α6β4 integrin is a transmembrane protein found in hemidesmosomes. As a heterodimer molecule containing two polypeptide chains its extracellular domain enters the basal lamina and interacts with type IV collagen suprastructure containing laminins (laminin-5), entactin/nidongen or the perlecan. on the extracellular surface of the hemidesmosome, laminin-5 molecules form threadlike anchoring filaments that extend from the integrin molecules to the structure of the basement membrane of epithelial adhesion. Mutation of the genes encoding laminin-5 chains results in junctional epidermolysis bullosa.[4]
Diagnosis
Classification
| OMIM | Name | Locus | Gene |
|---|---|---|---|
| 226730 | Junctional epidermolysis bullosa with pyloric atresia | 17q11-qter, 2q31.1 | ITGB4, ITGA6 |
| 226700 | Junctional epidermolysis bullosa, Herlitz type | 18q11.2, 1q32, 1q25-q31 | LAMA3, LAMB3, LAMC2 |
| 226650 | epidermolysis bullosa, junctional, non-Herlitz types (Generalized atrophic benign epidermolysis bullosa, Mitis junctional epidermolysis bullosa) | 18q11.2, 1q32, 17q11-qter, 1q25-q31, 10q24.3 | LAMA3, LAMB3, LAMC2, COL17A1, ITGB4 |
Junctional epidermolysis bullosa with pyloric atresia
Junctional epidermolysis bullosa with pyloric atresia is a rare autosomal recessive form of junctional epidermolysis bullosa that presents at birth with severe mucocutaneous fragility and gastric outlet obstruction.[5][6]: 557 It can be associated with ITGB4 or ITGA6.[7] This condition is also known as Carmi syndrome.
This condition is rare with ~100 cases reported in the literature.[8]
Herlitz type
Junctional epidermolysis bullosa gravis (also known as "Herlitz disease", "Herlitz syndrome", and "Lethal junctional epidermolysis bullosa") is the most lethal type of epidermolysis bullosa, a skin condition in which most patients do not survive infancy, characterized by blistering at birth with severe and clinically distinctive periorificial granulation tissue.[1]: 599 [6]: 557 [9]
JEB-H is generally caused by mutations in one of the three laminin-332 coding genes: LAMA3 (18q11.2), LAMB3 (1q32) and LAMC2 (1q25-q31).
Non-Herlitz type
These include:
- Generalized atrophic benign epidermolysis bullosa is a skin condition that is characterized by onset at birth, generalized blisters and atrophy, mucosal involvement, and thickened, dystrophic, or absent nails.[1]: 600 [6]: 557
- Mitis junctional epidermolysis bullosa (also known as "Nonlethal junctional epidermolysis bullosa") is a skin condition characterized by scalp and nail lesions, also associated with periorificial nonhealing erosions.[1]: 599 Mitis junctional epidermolysis bullosa is most commonly seen in children between the ages of 4 and 10 years old.[1]: 600
- Cicatricial junctional epidermolysis bullosa is a skin condition characterized by blisters that heal with scarring.[6]: 557 It was characterized in 1985.[10]
Treatment
In 2015, an Italian team of scientists, led by Michele De Luca at the University of Modena, successfully treated a seven-year-old Syrian boy who had lost 80% of his skin. The boy's family had fled Syria for Germany in 2013. Upon seeking treatment in Germany, he had lost the epidermis from almost his entire body, with only his head and a patch on his left leg remaining. The group of Italian scientists had previously pioneered a technique to regenerate healthy skin in the laboratory. They used this treatment on the boy by taking a sample from his remaining healthy skin and then genetically modifying the skin cells, using a virus to deliver a healthy version of the LAMB3 gene into the nuclei. The patient underwent two operations in autumn 2015, where the new epidermis was attached. The graft had integrated into the lower layers of skin within a month, and the modified epidermal stem cells sustained this transgenic epidermis, curing the boy.[11] The introduction of genetic changes could increase the chances of skin cancer in other patients, but if the treatment is deemed safe in the long term, scientists believe the approach could be used to treat other skin disorders.[12]
The use of gentamicin has been shown to provide some attenuation of this disease.[13]
See also
- Epidermolysis bullosa
- Junctional epidermolysis bullosa (veterinary medicine)
- Skin lesion
References
- 1 2 3 4 5 Freedberg IM, Fitzpatrick TB (2003). Fitzpatrick's dermatology in general medicine (6th ed.). New York, NY: McGraw-Hill. ISBN 978-0-07-138076-8.
- ↑ Bardhan, Ajoy; Bruckner-Tuderman, Leena; Chapple, Iain L. C.; Fine, Jo-David; Harper, Natasha; Has, Cristina; Magin, Thomas M.; Marinkovich, M. Peter; Marshall, John F.; McGrath, John A.; Mellerio, Jemima E. (2020-09-24). "Epidermolysis bullosa". Nature Reviews Disease Primers. 6 (1): 78. doi:10.1038/s41572-020-0210-0. ISSN 2056-676X. PMID 32973163. S2CID 221861310. Archived from the original on 2020-11-18. Retrieved 2022-03-31.
- ↑ "junctional epidermolysis bullosa". Genetics Home Reference. Archived from the original on 2017-10-04. Retrieved 2017-10-04.
- ↑ Ross MH, Pawlina W. Histology A Text And Atlas (7th ed.). LWW. ISBN 978-1-4698-8931-3.
- ↑ Todd-Sanford clinical diagnosis by laboratory methods, edited by Israel Davidsohn [and] John Bernard Henry (14th ed.). Philadelphia: Saunders. 1969. ISBN 978-0-7216-2921-6.
- 1 2 3 4 James WD, Andrews GE, Berger TG, Elston DM (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Philadelphia: Saunders. ISBN 978-0-7216-2921-6.
- ↑ Online Mendelian Inheritance in Man (OMIM): Epidermolysis bullosa junctionalis with pyloric atresia - 226730
- ↑ Mylonas KS, Hayes M, Ko LN, Griggs CL, Kroshinsky D, Masiakos PT (May 2018). "Clinical outcomes and molecular profile of patients with Carmi syndrome: A systematic review and evidence quality assessment". Journal of Pediatric Surgery. 54 (7): 1351–1358. doi:10.1016/j.jpedsurg.2018.05.019. PMID 29935895.
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ↑ Haber RM, Hanna W, Ramsay CA, Boxall LB (May 1985). "Cicatricial junctional epidermolysis bullosa". Journal of the American Academy of Dermatology. 12 (5 Pt 1): 836–44. doi:10.1016/S0190-9622(85)70105-3. PMID 4008687.
- ↑ Hirsch T, Rothoeft T, Teig N, Bauer JW, Pellegrini G, De Rosa L, et al. (November 2017). "Regeneration of the entire human epidermis using transgenic stem cells". Nature. 551 (7680): 327–332. Bibcode:2017Natur.551..327H. doi:10.1038/nature24487. PMC 6283270. PMID 29144448.
- ↑ Devlin, Hannah (2017-11-08). "Scientists grow replacement skin for boy suffering devastating genetic disorder". The Guardian. Archived from the original on 2017-11-09. Retrieved 2017-11-09.
- ↑ Hammersen J, Neuner A, Wild F, Schneider H (2019) Attenuation of Severe Generalized Junctional epidermolysis bullosa by systemic treatment with gentamicin. Dermatology 1-8
External links
| Classification | |
|---|---|
| External resources |
|
- GeneReview/NIH/UW entry on Junctional Epidermolysis Bullosa Archived 2022-01-20 at the Wayback Machine