Knobloch syndrome
| Knobloch syndrome | |
|---|---|
| Other names: Myopia retinal detachment encephalocele [1] | |
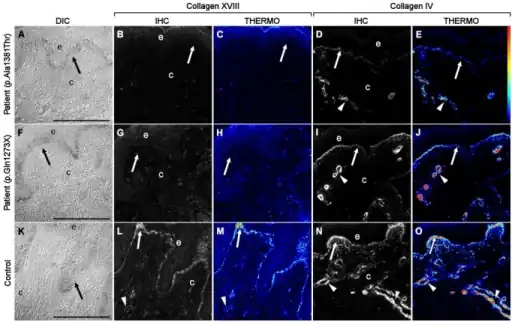 | |
| Immunofluorescent localization of type XVIII collagen in Knobloch syndrome (KS) and control skin samples. Differential interference contrast (DIC) images (A, F, K) show that the epithelial and connective tissues obtained from skin biopsies were intact and well preserved after being prepared using cryofixation and cryosubstitution techniques. Immunolocalization analyses of KS people carrying the p.A1381T change (B, C) and the nonsense mutation c.3277C>T (NC11–493 isoform position p.G1273X, GenBank AF018081.1; G, H) were negative for expression of collagen XVIII as compared to controls (L, M). Pseudocolor, thermo images showed that collagen XVIII expression was present in control samples (M) but undetectable in KS samples (C, H). Although the distribution pattern of type IV collagen was similar in both control (N, O), and KS (D, E, I, J) groups, the immunostaining intensity was noticeably lower in KS samples. The thermo color bar to the right of panel E indicates immunofluorescence staining intensity: red, higher levels; blue/black, lower levels. Arrows point to basement membrane, and arrowheads mark blood vessels. | |
Knobloch syndrome is a rare genetic disorder presenting severe eyesight problems and often a defect in the skull. It was named after the ophthalmologist William Hunter Knobloch (1926 - 2005),[2] who first described the syndrome in 1971.[3] A usual occurrence is a degeneration of the vitreous humour and the retina, two components of the eye. This breakdown often results in the separation of the retina (the light-sensitive tissue at the back of the eye) from the eye, called retinal detachment, which can be recurrent.[4] Extreme myopia (near-sightedness) is a common feature.[4] The limited evidence available from electroretinography suggests that a cone-rod pattern of dysfunction is also a feature.[5]
Knobloch syndrome is caused by mutations in an autosomal recessive inherited gene. These mutations have been found in the COL18A1 gene that instructs for the formation of a protein that builds collagen XVIII. This type of collagen is found in the basement membranes of various body tissues. Its deficiency in the eye is thought to be responsible for affecting normal eye development.[4] There are two types of Knobloch syndrome and the case has been made for a third.[6] When caused by mutations in the COL18A1 gene it is called Knobloch syndrome type 1.[7] The genes causing types II and III have yet to be identified.
Knobloch syndrome is also characterised by cataracts, dislocated lens with skull defects such as occipital encephalocele and occipital aplasia.[8] Encephalocele is a neural tube defect where the skull has not completely closed and sac-like protrusions of the brain can push through the skull; (it can also result from other causes).[9] In Knobloch's syndrome this is usually seen in the occipital region, and aplasia is the underdevelopment of tissue again in this reference in the occipital area.
References
- ↑ "Knobloch syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 19 September 2020. Retrieved 17 March 2019.
- ↑ Ramsay RC. William H. Knobloch, MD (1926-2005). Obituary. Arch Ophthalmol 2006; 124: 431; https://jamanetwork.com/journals/jamaophthalmology/fullarticle/417580 Archived 2018-06-16 at the Wayback Machine
- ↑ Knobloch WH, Layer JM. Retinal detachment and encephalocele. J Pediat Ophthalmol 1971; 8: 181-184
- 1 2 3 Reference, Genetics Home. "Knobloch syndrome". Genetics Home Reference. Archived from the original on 2020-08-14. Retrieved 2021-05-06.
- ↑ Hull, S; Arno, G; Ku, CA; Ge, Z; Waseem, N; Chandra, A; Webster, AR; Robson, AG; Michaelides, M; Weleber, RG; Davagnanam, I; Chen, R; Holder, GE; Pennesi, ME; Moore, AT (2016). "Molecular and Clinical Findings in Patients with Knobloch Syndrome" (PDF). JAMA Ophthalmol. 134 (7): 753–62. doi:10.1001/jamaophthalmol.2016.1073. PMID 27259167. Archived (PDF) from the original on 2018-07-24. Retrieved 2021-05-06.
- ↑ Khaliq, S; Abid, A; White, DR; Johnson, CA; Ismail, M; Khan, A; Ayub, Q; Sultana, S; Maher, ER; Mehdi, SQ (2007). "Mapping of a novel type III variant of Knobloch syndrome (KNO3) to chromosome 17q11.2". Am J Med Genet A. 143A (23): 2768–74. doi:10.1002/ajmg.a.31739. PMID 17975799. S2CID 28946327.
- ↑ "OMIM Entry - # 267750 - Knobloch syndrome 1; KNO1". www.omim.org. Archived from the original on 5 May 2017. Retrieved 24 February 2018.
- ↑ Aldahmesh, M. A.; Khan, A. O.; Mohamed, J. Y.; Alkuraya, H.; Ahmed, H.; Bobis, S.; Al-Mesfer, S.; Alkuraya, F. S. (2011). "Identification of ADAMTS18 as a gene mutated in Knobloch syndrome". J. Med. Genet. 48 (9): 597–601. doi:10.1136/jmedgenet-2011-100306. PMID 21862674. S2CID 22473497.
- ↑ Dorland's (2012). Dorland's Illustrated Medical Dictionary (32nd ed.). Elsevier Saunders. p. 613. ISBN 978-1-4160-6257-8.
Template:Congenital malformations and deformations of eye