Surfactant metabolism dysfunction
| Surfactant metabolism dysfunction | |
|---|---|
| Other names: Primary interstitial lung disease specific to childhood due to pulmonary surfactant protein anomalies | |
| Specialty | Pulmonology |
Surfactant metabolism dysfunction is a condition where pulmonary surfactant is insufficient for adequate respiration. Surface tension at the liquid-air interphase in the alveoli makes the air sacs prone to collapsing post expiration. This is due to the fact that water molecules in the liquid-air surface of alveoli are more attracted to one another than they are to molecules in the air. For sphere-like structures like alveoli, water molecules line the inner walls of the air sacs and stick tightly together through hydrogen bonds. These intermolecular forces put great restraint on the inner walls of the air sac, tighten the surface all together, and unyielding to stretch for inhalation. Thus, without something to alleviate this surface tension, alveoli can collapse and cannot be filled up again. Surfactant is essential mixture that is released into the air-facing surface of inner walls of air sacs to lessen the strength of surface tension. This mixture inserts itself among water molecules and breaks up hydrogen bonds that hold the tension.[1] Multiple lung diseases, like ISD or RDS, in newborns and late-onsets cases have been linked to dysfunction of surfactant metabolism.
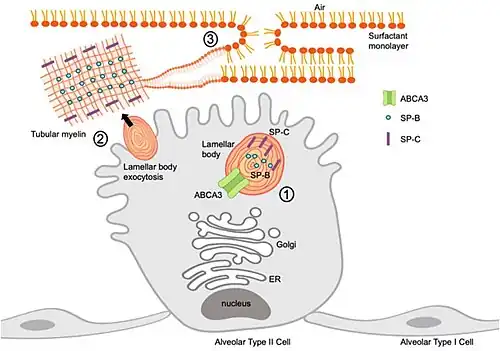
Surfactant is a mixture of 90% phospholipids and 10% other proteins, produced by epithelial type II cells in the alveolar. This mixture is made and packaged into lysosomally- derived structures called lamellar bodies. Lamellar bodies are then secreted into the liquid-air interphase surface of alveolar through membrane fusion initiated by influx of Ca2+.[2] Released pulmonary surfactant acts as a protective layer to prevent alveolar from collapsing due to surface tension. Furthermore, surfactants also contains some innate immune components to defend against pulmonary infections. Surfactant is classified into two types of proteins, hydrophilic proteins that are responsible for innate immune system, and hydrophobic proteins that carry out physical functions of pulmonary surfactant.[3] Surfactant metabolism dysfunction involves mutations or malfunctions of those hydrophobic proteins that lead to ineffective surfactant layer to protect alveolus integrity.[3] SP-B and SP-C are the two hydrophobic surfactant proteins that participate in its physical functions; these proteins are encoded by SFTPB and SFTPC genes on chromosomes 2 and 8 respectively.[4] Thus, mutations on these genes produce incomplete or nonfunctioning SP-B and SP-C proteins and lead to lung diseases.
Both SP-B and SP-C are synthesized in epithelial type II cells as large precursor proteins (proSP-B and proSP-C) and subsequently cleaved by proteolytic enzymes at both amino and carboxyl termini to produce functional mature proteins.[3] proSP-B and proSP-C are first made in the endoplasmic reticulum of epithelial type II cell, they are then translocated through Golgi apparatus to multivesicular bodies for delivery to lamellar bodies. During this transition, proteolytic processing begins to cleave precursor proteins. Once multivescular body reaches the membrane of lamellar body, both membranes fuse together so that processed proteins can be transported into lamellar body, where last steps of maturation for both SP-B and SP-C occur.[4] When lamellar body is ready to be secreted, exocytosis is initiated through influx of Ca2+, and lamellar membrane fuses with plasma membrane to release surfactant phospholipid contents into the surface of the cell.[2] SP-B and SP-C are responsible to carry out adsorption of the lipid monolayer at the liquid-air interphase to prevent post expiration atelectasis. Used surfactant phospholipid materials are taken up into epithelial type II cells by pulmonary macrophages.[2]
Another important protein that contributes to outcome of surfactant metabolism dysfunction is ABCA3, a transmembrane phospholipid transporter in lamellar body. ABCA3 has two ATP binding sites in the cytoplasmic domain to power phospholipid transportation through ATP hydrolysis. ABCA3 is synthesized in endoplasmic reticulum and transported through Golgi apparatus to the membrane of lamellar body.[4] Once inserted into the membrane, ABCA3 can help deliver surfactant lipids into the lumen of lamellar body, and create tightly packed internal environment of surfactant lipids and surfactant proteins. Mutations in ABCA3 cause failure in lamellar body synthesis and result in decreased production of surfactant, along with deficiency of SP-B and SP-C.[3]
Types
| Type | OMIM | Gene | Locus |
|---|---|---|---|
| SMDP1 | 265120 | SFTPB | 2p12 |
| SMDP2 | 610913 | SPTPC | 8p21 |
| SMDP3 | 610921 | ABCA3 | 16p13 |
| SMDP4 | 300770 | CSF2RA | Xp |
Signs and symptoms
In terms of the presentation of Surfactant metabolism dysfunction we find the following:[5]
Cause
Surfactant metabolism dysfunction describes a group of dysfunctions caused by different mutations in surfactant related genes. Severe deficiency of pulmonary surfactant due to disturbed metabolism of any of these proteins can lead to some form of interstitial lung disease in newborns and adults. These conditions share similar pathophysiology and overlapping phenotypes because surfactant gene products interactively communicate and control one another.[3] Thus, dysfunction of a surfactant protein, or relating protein, generates deficiencies of others.
SFTPB mutations
Most disease-causing mutations in SFTPB result in a complete lack of mature SP-B protein 265120. Lung disease is inherited in an autosomal recessive manner, requiring mutations in both alleles. Surfactant produced by infants with SP-B deficiency is abnormal in composition and does not function normally in lowering surface tension.
More than 40 different mutations along the length of SFTPB gene have been accounted for in surfactant metabolism dysfunction. SFTPB mutations are inherited in autosomal recessive fashion, loss-of-function mutation on both alleles are required for full expression of disease. About 2/3 or 60%-70% of those accounted disease-causing alleles come from a frameshift mutation, called 121ins2, on exon 4 of SFTPB gene, which also accounts for ~65% of US cases. The rest of the mutated alleles come from nonsense, missense, splice-site mutations, and other possible insertion and deletion mutations throughout the entire gene.[4] These mutations cause total absence or loss-of-function of SP-B and lead to imbalance in surfactant homeostasis. Since SP-B has a major role in surfactant biogenesis and spreading of surfactant and lipid layer, any disruption to existence of SP-B results in ineffective respiration and lethal pulmonary conditions at birth.[7] Pathology manifestation in full-term infant resembles characteristics of newborn with Respiratory Distress Syndrome.[8] Imaging of epithelial type II cells with SP-B deficiency shows immature lamellar bodies without tightly packed membranes, but rather with loose and unorganized membranes. The ratio of phospholipid-protein also decreases with abnormal phospholipids. In addition, surfactant collected from SP-B deficiency epithelial type II cells is not as effective in lowering surface tension and creating film as normal surfactant.[3] Immunohistochemical features of SP-B deficiency show decreased levels of proSP-B and SP-B proteins, along with increased presence of immuno protein SP-A and partially processed intermediate peptides of proSP-C.[4] Appearance of partially processed proSP-C shows significance of mature SP-B in biogenesis and processing of SP-C. Absences of both proSP-B and SP-B proteins are observed in frameshift and nonsense mutations of SFTPB, while low level of proSP-B is detected in missense, in-frame deletionof insertion mutations. However, these mutations prevent proSP-B from fully mature into SP-B, resulting in deficiency of SP-B and surfactant.
SFTPC mutations
Familial cases of SP-C dysfunction 610913 are inherited in an autosomal dominant pattern, although the onset and severity of lung disease are highly variable, even within the same family.
More than 40 distinct mutation variations in SFTPC gene have also been described in patients. Wild-type SP-C proteins are embedded inside the phospholipid bilayer of epithelial type II cell and function to generate and maintain monolayer of surfactant on alveolar surface.[4] Individuals with mutated SFTPC genes tend to manifest lung diseases in late childhood or adulthood. Mutated alleles are inherited in autosomal dominant fashion, although de novo mutations can also cause sporadic emergence of diseases. The age of onset and severity vary significantly among patients with SFTPC mutations, some only manifest symptoms in fifth or sixth decade.[3] Most of these mutations are missense, but there have been recordings of frameshift, splice-site mutations, together with small insertions or deletions along the carboxyl terminal of SFTPC. Mutations in SFTPC gene are thought to prevent proSP-C peptides from being fully processed into mature SP-C proteins. ProSP-C proteins tend to self-accumulate along the secretory pathway, due to high hydrophobic nature, and may activate cellular destruction response. SFTPC mutations cause proSP-C proteins to aggregate and misfold during secretory process.[3] These folded proteins trigger unfolded protein response (UPR) and cellular apoptosis to get rid of clusters of mutated peptides. Patients with SP-C dysfunction show lack of mature SP-C in epithelial type II cells and up-regulation of UPR.[4] SFTPC mutation with highest occurrence frequency is substitution of threonine for isoleucine in codon 73, termed I73T, found in more than 25% of patients with SP-C related disorders. Staining of proSP-C shows diffuse staining strictly in cytoplasm and accumulation of immunoreactive substances surrounding the nucleus.[4] Evaluation of diseases related to SFTPC mutations show association with chronic parenchymal lung disease.
ABCA3 mutations
Mutations in ABCA3 appear to be the most common cause of genetic surfactant dysfunction in humans.[9][10][11] The mutations result in a loss of or reduced function of the ABCA3 protein, and are inherited in an autosomal recessive manner 610921.
There are more than 150 different mutations throughout ABCA3 gene with various allelic heterogeneity, making it the biggest class of genetic cause of surfactant dysfunction. Like SP-B deficiency, ABCA3 mutations are inherited in autosomal recessive trait. Mutations of ABCA3 consist of missense, nonsense, frameshift, splice-cite, insertion or deletion.[4] These mutations are classified into two types of ABCA3 mutations, those that preclude normal trafficking of ABCA3 from ER to lamellar membrane, and those that affect ATP-binding ability of ABCA3 needed for phospholipid transportation.[3] Due to its roles in lamellar body biogenesis and maturation of surfactant proteins, epithelial type II cells with altered ABCA3 exhibit premature lamellar bodies and damaged maturation of SP-B/SP-C. Surfactant samples from patients with ABCA3 deficiency do not lower surface tension as effectively. Affected surface tension ability results from incomplete formation of lamellar bodies, due to lack of lipid influx by ABCA3. Immunostaining of SP-B in ABCA3 patients show decreased level of mature SP-B and impaired process of proSP-B to SP-B, thus, confirming why ABCA3 dysfunction leads to severe surfactant metabolism dysfunction.[4]
Diagnosis
Non-invasive genetic testing can be used to infer possible interstitial lung disorders caused by surfactant metabolism dysfunction. Although these sequencing tests can take up to several weeks, which may not be so useful in case of acute respiratory problems in newborns. Overlapping phenotypes of surfactant metabolism dysfunction and other interstitial lung diseases make it hard to propose definitive diagnosis for surfactant disorders. Overall testings, family history, external factors, and clinical presentations should all be considered to diagnose surfactant metabolism dysfunction. Testing for surfactant metabolism dysfunction should be considered for newborns with diffuse lung disease or hypoxemia, especially in families with history of neonatal lung diseases or ILD in adults. Neonatal and adult onset lung diseases with unfound causes should also be tested early for surfactant dysfunction.[3] ABCA3 and SP-B dysfunctions manifest in newborns and progress aggressively within the first few months of life, thus, testing for ABCA3 and SP-B disorders should preclude those for SP-C, especially when infants are showing symptoms of ILD or diffuse lung disease. Distinctions between SP-B and ABCA3 are ABCA3 tends to occur in families with neonatal lung disease history, and SP-B testing almost unneeded in older children.[3] Late on-set conditions with inheritance in dominant fashion should infer SP-C dysfunction. Antibodies against proSP-B, proSP-C, SP-B, SP-C, and ABCA3 have been thoroughly developed, which makes detection for these proteins highly accessible and accurate.[4] Immuno staining of each of these type of surfactant dysfunction differs in absence and presence of specific proteins and propeptides, thus immunohistochemisty can help decipher which type of deficiency is being dealt with. In addition, hypothyroidism can cause damaged production of NKX2.1 proteins, which can lead to insufficient transcription of multiple surfactant proteins.
Treatment
Neonates with surfactant metabolism dysfunctions, especially those with SP-B disorder, only have lung transplantation as one possible choice of treatment.[3] Children with lung transplant due to surfactant metabolism dysfunction perform on similar level to those with transplant for due to other reasons.[3] Some less severe cases of ABCA3 dysfunctions manifest in late childhood or adult hood are due to missense mutations that result in semi-sufficient levels of active surfactant, while SP-C clinical presentation varies greatly depending on level of penetration of the mutated alleles.[4]
See also
References
- ↑ Seadler, Benjamin D.; Kaushik, Ravi; Sharma, Sandeep (2020), "Physiology, Alveolar Tension", StatPearls, StatPearls Publishing, PMID 30969647, archived from the original on 2023-10-26, retrieved 2020-04-24
- 1 2 3 Olmeda, Bárbara; Martínez-Calle, Marta; Pérez-Gil, Jesus (2017-01-01). "Pulmonary surfactant metabolism in the alveolar airspace: Biogenesis, extracellular conversions, recycling". Annals of Anatomy - Anatomischer Anzeiger. 209: 78–92. doi:10.1016/j.aanat.2016.09.008. ISSN 0940-9602. PMID 27773772. Archived from the original on 2023-10-26. Retrieved 2023-04-16.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Gower, W. Adam; Nogee, Lawrence M. (December 2011). "Surfactant Dysfunction". Paediatric Respiratory Reviews. 12 (4): 223–229. doi:10.1016/j.prrv.2011.01.005. ISSN 1526-0542. PMC 3201772. PMID 22018035.
- 1 2 3 4 5 6 7 8 9 10 11 12 Wert, Susan E.; Whitsett, Jeffrey A.; Nogee, Lawrence M. (2009). "Genetic Disorders of Surfactant Dysfunction". Pediatric and Developmental Pathology. 12 (4): 253–274. doi:10.2350/09-01-0586.1. ISSN 1093-5266. PMC 2987676. PMID 19220077.
- ↑ "Surfactant dysfunction: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 2 August 2023. Retrieved 25 October 2023.
- ↑ Cooney, Ashley L.; Wambach, Jennifer A.; Sinn, Patrick L.; McCray, Paul B. (2022). "Gene Therapy Potential for Genetic Disorders of Surfactant Dysfunction". Frontiers in Genome Editing. 3. doi:10.3389/fgeed.2021.785829/full. ISSN 2673-3439.
- ↑ Hawgood, Samuel; Derrick, Matthew; Poulain, Francis (1998-11-19). "Structure and properties of surfactant protein B". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1408 (2): 150–160. doi:10.1016/S0925-4439(98)00064-7. ISSN 0925-4439. PMID 9813296.
- ↑ McFetridge, Lynne; McMorrow, Aoife; Morrison, Patrick J; Shields, Michael D (January 2009). "Surfactant Metabolism Dysfunction and Childhood Interstitial Lung Disease (chILD)". The Ulster Medical Journal. 78 (1): 7–9. ISSN 0041-6193. PMC 2629012. PMID 19252722.
- ↑ Brasch, F; Griese, M; Tredano, M; Johnen, G; Ochs, M; Rieger, C; Mulugeta, S; Müller, KM; Bahuau, M; Beers, MF (Jul 2004). "Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene". The European Respiratory Journal. 24 (1): 30–9. doi:10.1183/09031936.04.00000104. PMID 15293602.
- ↑ Shulenin, S; Nogee, LM; Annilo, T; Wert, SE; Whitsett, JA; Dean, M (Mar 25, 2004). "ABCA3 gene mutations in newborns with fatal surfactant deficiency". The New England Journal of Medicine. 350 (13): 1296–303. doi:10.1056/NEJMoa032178. PMID 15044640.
- ↑ Somaschini, M; Nogee, LM; Sassi, I; Danhaive, O; Presi, S; Boldrini, R; Montrasio, C; Ferrari, M; Wert, SE; Carrera, P (Jun 2007). "Unexplained neonatal respiratory distress due to congenital surfactant deficiency". The Journal of Pediatrics. 150 (6): 649–53, 653.e1. doi:10.1016/j.jpeds.2007.03.008. PMID 17517255.
Further reading
- Reference, Genetics Home. "surfactant dysfunction". Genetics Home Reference. Archived from the original on 7 November 2017. Retrieved 5 November 2017.
External links
| External resources |
|
|---|