X-linked intellectual disability
| X-linked intellectual disability | |
|---|---|
| Other names: X-linked mental retardation | |
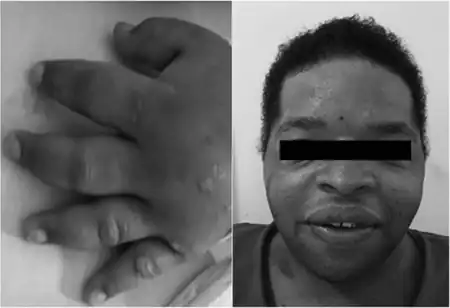 | |
| Images of the dysmorphology examination typical of Coffin-Lowry syndrome | |
X-linked intellectual disability refers to medical disorders associated with X-linked recessive inheritance that result in intellectual disability.
As with most X-linked disorders, males are more heavily affected than females.[1] Females with one affected X chromosome and one normal X chromosome tend to have milder symptoms.
Unlike many other types of intellectual disability, the genetics of these conditions are relatively well understood.[2][3] It has been estimated there are ~200 genes involved in this syndrome; of these ~100 have been identified.[4] Many of these genes are found on the short 'p' arm of the chromosome, and duplications at Xp11.2 are associated with the syndromic form of the condition.[5][6]
X-linked intellectual disability accounts for ~16% of all cases of intellectual disability in males.[7]
Syndromes
Several X-linked syndromes include intellectual disability as part of the presentation. These include:
Genes
Following is a list of genes located on the X chromosome and linked to intellectual disability. There are also several loci that have not been associated with a specific gene.
- IQSEC2: encodes an exchange factor for the Arf family of small GTP binding proteins, involved in the formation of secretory vesicles.[8]
- TM4SF2: is a member of the 4 transmembrane domains family of proteins (tetraspanins, see TSPAN7). This gene is also associated with neuropsychiatric diseases such as Huntington's chorea.[9]
- AP1S2: AP-1 complex subunit sigma-2.[10][11] Adaptor protein complex 1 is found on the cytoplasmic face of vesicles located at the Golgi complex, where it mediates both the recruitment of clathrin to the membrane and the recognition of sorting signals within the cytosolic tails of transmembrane receptors.
- ACSL4: Long-chain-fatty-acid—CoA ligase 4 is an enzyme of the long-chain fatty-acid-coenzyme A ligase family. It converts free long-chain fatty acids into fatty acyl-CoA esters, and thereby play a key role in lipid biosynthesis and fatty acid degradation.[12] This isozyme preferentially utilizes arachidonate as substrate.
- ZNF41: Zinc finger protein 41 is a likely zinc finger family transcription factor.[13]
- DLG3: Disks large homolog 3, also named neuroendocrine-DLG or synapse-associated protein 102 (SAP-102).[14] DLG3 is a member of the membrane-associated guanylate kinase (MAGUK) superfamily.
- FTSJ1: Transfert RNA methyltransferase 1 is a member of the S-adenosylmethionine-binding protein family. This nucleolar protein is involved in the processing and modification of tRNA.[15][16]
- GDI1: RabGDI alpha makes a complex with geranylgeranylated small GTP-binding proteins of the Rab family and keeps them in the cytosol.
- MECP2: methyl CpG binding protein 2 is a transcription regulator, which represses transcription from methylated gene promoters. It appears to be essential for the normal function of nerve cells.[17] In contrast to other MBD family members, MECP2 is X-linked and subject to X inactivation. MECP2 gene mutations are the cause of most cases of Rett syndrome, a progressive neurologic developmental disorder and one of the most common causes of intellectual disability in women.
- ARX: Aristaless related homeobox, is a protein associated with intellectual disability and lissencephaly. This gene is a homeobox-containing gene expressed during development. The expressed protein contains two conserved domains, a C-peptide (or aristaless domain) and the prd-like class homeobox domain. It is a member of the group-II aristaless-related protein family whose members are expressed primarily in the central and/or peripheral nervous system. This gene is involved in CNS and pancreas development. Mutations in this gene cause X-linked intellectual disability and epilepsy.[18]
- KDM5C: Lysine-specific demethylase 5C is an enzyme that in humans is encoded by the KDM5C gene a member of the SMCY homolog family and encodes a protein with one ARID domain, one JmjC domain, one JmjN domain and two PHD-type zinc fingers. The DNA-binding motifs suggest this protein is involved in the regulation of transcription and chromatin remodeling.[19]
- PHF8: PHD finger protein 8 belongs to the family of ferrous iron and 2-oxoglutarate dependent oxygenases,[20] and is a histone lysine demethylase with selectivity for the di-and monomethyl states.[21]
- FMR2: Fragile mental retardation 2 (FMR2: synonym AFF2),[22] the protein belongs to the AFF family which currently has four members: AFF1/AF4, AFF2/FMR2, AFF3/LAF4 and AFF4/AF5q31.[23] All AFF proteins are localized in the nucleus and have a role as transcriptional activators with a positive action on RNA elongation. AFF2/FMR2, AFF3/LAF4 and AFF4/AF5q31 localize in nuclear speckles (subnuclear structures considered to be storage/modification sites of pre-mRNA splicing factors) and are able to bind RNA with a high apparent affinity for the G-quadruplex structure. They appear to modulate alternative splicing via the interaction with the G-quadruplex RNA-forming structure.
- Slc6a8: Creatine transporter is a protein that is required for creatine to enter the cell. Creatine is essential for maintaining ATP levels in cells with a high energy demand.[24]
- GSPT2[25]
- MAGED1[26]
- UBE2A[27]
See also
References
- ↑ "Fragile X Syndrome - X-linked Mental Retardation and Macroorchidism". International Birth Defect Information Systems. Archived from the original on 2010-11-23. Retrieved 2010-12-10.
- ↑ Ropers HH, Hamel BC (January 2005). "X-linked mental retardation". Nature Reviews. Genetics. 6 (1): 46–57. doi:10.1038/nrg1501. PMID 15630421. S2CID 427210.
- ↑ Lugtenberg D, Veltman JA, van Bokhoven H (September 2007). "High-resolution genomic microarrays for X-linked mental retardation". Genetics in Medicine. 9 (9): 560–565. doi:10.1097/GIM.0b013e318149e647. PMID 17873643.
- ↑ Stevenson RE, Schwartz CE (2009). "X-linked intellectual disability: unique vulnerability of the male genome". Developmental Disabilities Research Reviews. 15 (4): 361–368. doi:10.1002/ddrr.81. PMID 20014364.
- ↑ "OMIM Entry - # 300705 - CHROMOSOME Xp11.22 DUPLICATION SYNDROME". omim.org. Archived from the original on 2017-05-06. Retrieved 2018-03-09.
- ↑ "Microduplication Xp11.22-p11.23 syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2021-03-21. Retrieved 2018-03-09.
- ↑ Stevenson RE, Schwartz CE (2009). "X-linked intellectual disability: unique vulnerability of the male genome". Developmental Disabilities Research Reviews. 15 (4): 361–368. doi:10.1002/ddrr.81. PMID 20014364.
- ↑ Shoubridge C, Tarpey PS, Abidi F, Ramsden SL, Rujirabanjerd S, Murphy JA, et al. (June 2010). "Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability". Nature Genetics. 42 (6): 486–488. doi:10.1038/ng.588. PMC 3632837. PMID 20473311.
- ↑ Abidi FE, Holinski-Feder E, Rittinger O, Kooy F, Lubs HA, Stevenson RE, Schwartz CE (June 2002). "A novel 2 bp deletion in the TM4SF2 gene is associated with MRX58". Journal of Medical Genetics. 39 (6): 430–433. doi:10.1136/jmg.39.6.430. PMC 1735161. PMID 12070254.
- ↑ Tarpey PS, Stevens C, Teague J, Edkins S, O'Meara S, Avis T, et al. (December 2006). "Mutations in the gene encoding the Sigma 2 subunit of the adaptor protein 1 complex, AP1S2, cause X-linked mental retardation". American Journal of Human Genetics. 79 (6): 1119–1124. doi:10.1086/510137. PMC 1698718. PMID 17186471.
- ↑ "AP1S2 adaptor-related protein complex 1, sigma 2 subunit". Entrez Gene. National Center for Biotechnology Information, U.S. National Library of Medicine. Archived from the original on 2022-12-18. Retrieved 2022-09-10.
- ↑ Piccini M, Vitelli F, Bruttini M, Pober BR, Jonsson JJ, Villanova M, et al. (February 1998). "FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation". Genomics. 47 (3): 350–358. doi:10.1006/geno.1997.5104. PMID 9480748.
- ↑ Franzè A, Archidiacono N, Rocchi M, Marino M, Grimaldi G (April 1991). "Isolation and expression analysis of a human zinc finger gene (ZNF41) located on the short arm of the X chromosome". Genomics. 9 (4): 728–736. doi:10.1016/0888-7543(91)90367-N. PMID 2037297.
- ↑ Stathakis DG, Lee D, Bryant PJ (April 1998). "DLG3, the gene encoding human neuroendocrine Dlg (NE-Dlg), is located within the 1.8-Mb dystonia-parkinsonism region at Xq13.1". Genomics. 49 (2): 310–313. doi:10.1006/geno.1998.5243. PMID 9598320.
- ↑ Ramser J, Winnepenninckx B, Lenski C, Errijgers V, Platzer M, Schwartz CE, et al. (September 2004). "A splice site mutation in the methyltransferase gene FTSJ1 in Xp11.23 is associated with non-syndromic mental retardation in a large Belgian family (MRX9)". Journal of Medical Genetics. 41 (9): 679–683. doi:10.1136/jmg.2004.019000. PMC 1735884. PMID 15342698.
- ↑ Guy MP, Phizicky EM (January 2015). "Conservation of an intricate circuit for crucial modifications of the tRNAPhe anticodon loop in eukaryotes". RNA. 21 (1): 61–74. doi:10.1261/rna.047639.114. PMC 4274638. PMID 25404562.
- ↑ Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY (May 2008). "MeCP2, a key contributor to neurological disease, activates and represses transcription". Science. 320 (5880): 1224–1229. Bibcode:2008Sci...320.1224C. doi:10.1126/science.1153252. PMC 2443785. PMID 18511691.
- ↑ Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, et al. (April 2002). "ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation". Human Molecular Genetics. 11 (8): 981–991. doi:10.1093/hmg/11.8.981. PMID 11971879.
- ↑ Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, et al. (February 2005). "Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation". American Journal of Human Genetics. 76 (2): 227–236. doi:10.1086/427563. PMC 1196368. PMID 15586325.
- ↑ Loenarz C, Schofield CJ (March 2008). "Expanding chemical biology of 2-oxoglutarate oxygenases". Nature Chemical Biology. 4 (3): 152–156. doi:10.1038/nchembio0308-152. PMID 18277970.
- ↑ Loenarz C, Ge W, Coleman ML, Rose NR, Cooper CD, Klose RJ, et al. (January 2010). "PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nepsilon-dimethyl lysine demethylase". Human Molecular Genetics. 19 (2): 217–222. doi:10.1093/hmg/ddp480. PMC 4673897. PMID 19843542.
- ↑ Stettner GM, Shoukier M, Höger C, Brockmann K, Auber B (August 2011). "Familial intellectual disability and autistic behavior caused by a small FMR2 gene deletion". American Journal of Medical Genetics. Part A. 155A (8): 2003–2007. doi:10.1002/ajmg.a.34122. PMID 21739600. S2CID 9568277.
- ↑ Melko M, Douguet D, Bensaid M, Zongaro S, Verheggen C, Gecz J, Bardoni B (May 2011). "Functional characterization of the AFF (AF4/FMR2) family of RNA-binding proteins: insights into the molecular pathology of FRAXE intellectual disability". Human Molecular Genetics. 20 (10): 1873–1885. doi:10.1093/hmg/ddr069. PMID 21330300.
- ↑ Cecil KM, Salomons GS, Ball WS, Wong B, Chuck G, Verhoeven NM, et al. (March 2001). "Irreversible brain creatine deficiency with elevated serum and urine creatine: a creatine transporter defect?". Annals of Neurology. 49 (3): 401–404. doi:10.1002/ana.79. PMID 11261517. S2CID 38756630.
- ↑ Grau C, Starkovich M, Azamian MS, Xia F, Cheung SW, Evans P, et al. (2017). "Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability". PLOS ONE. 12 (4): e0175962. Bibcode:2017PLoSO..1275962G. doi:10.1371/journal.pone.0175962. PMC 5393878. PMID 28414775.
- ↑ Grau C, Starkovich M, Azamian MS, Xia F, Cheung SW, Evans P, et al. (2017). "Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability". PLOS ONE. 12 (4): e0175962. Bibcode:2017PLoSO..1275962G. doi:10.1371/journal.pone.0175962. PMC 5393878. PMID 28414775.
- ↑ Czeschik JC, Bauer P, Buiting K, Dufke C, Guillén-Navarro E, Johnson DS, et al. (September 2013). "X-linked intellectual disability type Nascimento is a clinically distinct, probably underdiagnosed entity". Orphanet Journal of Rare Diseases. 8: 146. doi:10.1186/1750-1172-8-146. PMC 4015352. PMID 24053514.
External links
| Classification |
|
|---|