Campomelic dysplasia
| Campomelic dysplasia | |
|---|---|
 | |
| This condition is inherited in an autosomal dominant manner | |
| Specialty | Medical genetics |
Campomelic dysplasia (CMD) is a rare genetic disorder characterized by bowing of the long bones and many other skeletal and extraskeletal features. It is frequently lethal in the neonatal period due to respiratory insufficiency, but the severity of the disease is variable, and some patients survive into adulthood. The name is derived from the Greek roots campo (or campto), meaning bent, and melia, meaning limb. An unusual aspect of the disease is that up to two-thirds of affected 46,XY genotypic males display a range of disorders of sexual development (DSD) and genital ambiguities or may even develop as normal phenotypic females as in complete 46 XY sex reversal. An atypical form of the disease with absence of bowed limbs is called, prosaically, acampomelic campomelic dysplasia (ACD) and is found in about 10% of patients, particularly those surviving the neonatal period.
Signs and symptoms
While the definitive presentation of the disease is a patient having bowed lower limbs and sex reversal in 46,XY males, there are other clinical criteria that can be used, absent these characteristics, to make the diagnosis. Patients may present with underdeveloped shoulder blades, shortened and angulated lower limbs, a vertically oriented and narrow pelvis, an enlarged head, an undersized jaw, cleft palate, flat nasal bridge, low set ears, club feet, dislocated hips, 11 pairs of ribs instead of 12, or bone abnormalities in the neck and spine. Respiratory distress can be caused by an underdeveloped trachea which collapses on inhalation or by insufficient rib cage development.
Genetics
CMD is caused by chromosomal abnormalities, generally spontaneously arising or de novo mutations, in or around the gene SOX9 on the long arm of chromosome 17, specifically at position 17q24. The SOX9 gene codes for a protein transcription factor which, when expressed at the embryonic stage, plays an important role in determining sexual characteristics and greatly influences skeletal development. When the SRY gene of the Y chromosome is expressed in human embryos, a cascade of gene interactions controlled by SOX9 begins and ultimately leads to male gender.
Numerous mutations have been identified involving the SOX9 gene that cause some form of CMD. Any mutation within the coding region of SOX9 can cause campomelic dysplasia and 75% of the reported mutations lead to sex reversal. Four major classes of heterozygous SOX9 mutations can cause CMD: amino acid substitutions in the HMG-box, truncations or frameshifts that alter the C-terminal end, mutations at the splice junction, and chromosomal translocations. Additionally, mutations upstream from SOX9 can also cause CMD. Several researchers have reported cis-acting control elements upstream of SOX9. Translocation breakpoints scattered over 1Mb proximal to SOX9 indicate the presence of an extended control region.
The lack of correlation between specific genetic mutations and observed phenotype, particularly with regard to sex reversal, give clear evidence of the variable expressivity of the disease.
Milder forms of the disease, seen in those who live beyond the neonatal period and those with ACD, may be attributable to somatic mosaicism—particularly for those with mutations within the SOX9 coding region. [1] Chromosomal aberrations in the upstream control regions or residual activity of the mutant SOX9 protein could also be responsible for the milder forms of the disease.[1] Long-term survivors of CMD are significantly more likely to have translocation and inversion mutations upstream of SOX9 rather than mutations in the SOX9 coding region itself.
Diagnosis
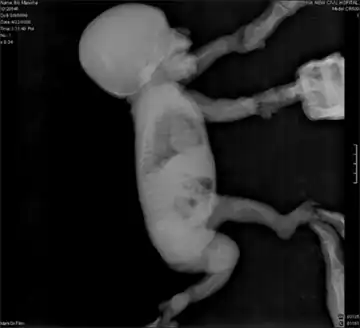
In utero sonographic diagnosis is possible when characteristic features such as bilateral bowed femurs and tibia, clubbed feet, prominent curvature of the neck, a bell-shaped chest, pelvic dilation, and/or an undersized jaw are apparent. [2] Radiographic techniques are generally used only postnatally and also rely on prototypical physical characteristics. [3]
Screening
Genetic screening is also typically done postnatally, including PCR typing of microsatellite DNA and STS markers as well as comparative genomic hybridization (CGH) studies using DNA microarrays.
In some cases PCR and sequencing of the entire SOX9 gene is used to diagnose CMD.
Many different translocation breakpoints and related chromosomal aberrations in patients with CMD have been identified.
Prognosis
In nearly 95% of the cases, death occurs in the neonatal period due to respiratory distress, generally related to small chest size or insufficient development of the trachea and other upper airway structures.[1]
Among survivors of CMD, the skeletal malformations change over time to include worsening scoliosis or kyphosis resulting in decreased trunk size relative to the limb length. Neurological damage is also often seen including intellectual disability and deafness. Even among survivors of the prenatal period, CMD patients have shortened life spans due to lifelong respiratory issues. Those patients with ambiguous genitalia or sex reversal at birth, of course, maintain that state, and are either sterile or have reduced fertility.
Epidemiology
Campomelic dysplasia has a reported incidence of 0.05-0.09 per 10000 live births.
References
- 1 2 3 S. Corbani; E. Chouery; B. Eid; et al. (2010). "Mild Campomelic Dysplasia: Report on a Case and Review". Mol Syndromol. 1 (4): 163–168. doi:10.1159/000322861. PMC 3042119. PMID 21373255.
- ↑ K. Eger (2005). "Campomelic Dysplasia". J Diag Medical Sonography. 21 (4): 345–349. doi:10.1177/8756479305278970.
- ↑ J. Goyal; A. Gupta; V. Shah (2011). "Campomelic dysplasia". Indian J Hum Genet. 17 (3): 247–248. doi:10.4103/0971-6866.92085. PMC 3277002. PMID 22346005.
External links
| Classification |
|
|---|---|
| External resources |
|
- GeneReviews/NCBI/NIH/UW entry on Campomelic Dysplasia Archived 2010-06-09 at the Wayback Machine