Holt–Oram syndrome
| Holt-Oram syndrome | |
|---|---|
| Other names: Heart-hand syndrome, Atrio-digital syndrome, Atriodigital dysplasia | |
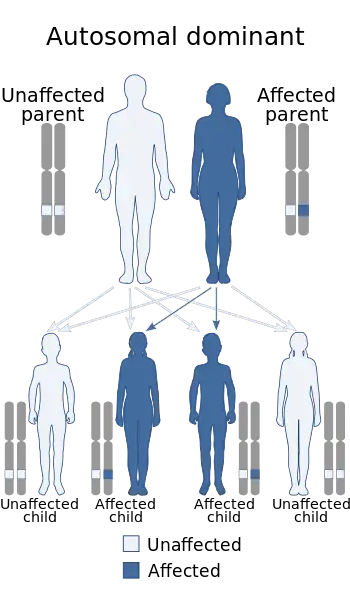 | |
| Holt-Oram syndrome has an autosomal dominant pattern of inheritance | |
Holt–Oram syndrome (also called atrio-digital syndrome, atriodigital dysplasia, cardiac-limb syndrome, heart-hand syndrome type 1, HOS, ventriculo-radial syndrome) is an autosomal dominant disorder that affects bones in the arms and hands (the upper limbs) and often causes heart problems.[1] The syndrome may include an absent radial bone in the forearm, an atrial septal defect in the heart, or heart block.[2] It affects approximately 1 in 100,000 people.[2]
Symptoms and signs
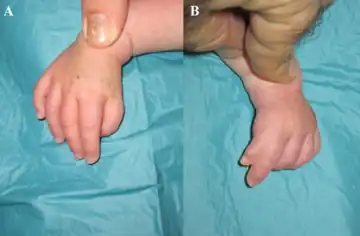
All people with Holt-Oram syndrome have, at least one, abnormal wrist bone, which can often only be detected by X-ray.[1] Other bone abnormalities are associated with the syndrome. These vary widely in severity, and include a missing thumb, a thumb that looks like a finger, upper arm bones of unequal length or underdeveloped, partial or complete absence of bones in the forearm, and abnormalities in the collar bone or shoulder blade.[1][2] Bone abnormalities may affect only one side of the body or both sides; if both sides are affected differently, the left side is usually affected more severely.[1]
About 75 percent of individuals with Holt–Oram syndrome also have congenital heart problems,[2] with the most common being defects in the tissue wall between the upper chambers of the heart (atrial septal defect) or the lower chambers of the heart (ventricular septal defect).[3] People with Holt–Oram syndrome may also have cardiac conduction disease, or abnormalities in the electrical system that coordinates contractions of the heart chambers.[1] Cardiac conduction disease can lead to slow heart rate (bradycardia); rapid, ineffective contraction of the heart muscles (fibrillation); and heart block.[1][2] People with Holt-Oram syndrome may have only congenital heart defects, only cardiac conduction disease, both or neither.[1][2]
Genetics
Mutations in the TBX5 gene cause Holt–Oram syndrome.[1] The TBX5 gene produces a protein that is critical for the proper development of the heart and upper limbs before birth.[1]
Holt–Oram syndrome has an autosomal dominant pattern of inheritance, meaning one abnormal copy of the gene is sufficient to cause disease, which each child has a 50% chance of inheriting from an affected parent.[1] However, in 85 percent of cases, the gene mutation isn't inherited, but a new mutation.[2]
Diagnosis
Diagnosis may be made on physical features alone, if a person has an arm or hand bone abnormality and a personal or family history of heart problems.[2] If the symptoms aren't enough to diagnose, a person may undergo genetic testing for the mutations associated with the syndrome.[2]
Treatment
A person with Holt-Oram syndrome may need various treatments, depending on how the syndrome manifests. Surgery, prosthetics and physical or occupational therapy can help people with bone abnormalities.[2] Heart defects may call for surgery, medication, pacemakers or close monitoring.[2] Pregnant women with Holt-Oram syndrome and heart abnormalities should be followed by a cardiologist during pregnancy.[2]
History
It is named for Mary Holt and Samuel Oram, who published a paper on it in 1960.[4][5]
See also
References
- 1 2 3 4 5 6 7 8 9 10 "Holt-Oram syndrome". Genetics Home Reference. U.S. National Library of Medicine. June 2014. Archived from the original on 26 October 2020. Retrieved 18 April 2018.
- 1 2 3 4 5 6 7 8 9 10 11 12 McDermott DA, Fong JC, Basson CT. Holt-Oram Syndrome. 2004 Jul 20 [Updated 2015 Oct 8]. In Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1111/
- ↑ Bossert, T; Walther, T; Gummert, J; Hubald, R; Kostelka, M; Mohr, FW (October 2002). "Cardiac malformations associated with the Holt-Oram syndrome—report on a family and review of the literature". The Thoracic and Cardiovascular Surgeon. 50 (5): 312–4. doi:10.1055/s-2002-34573. PMID 12375192.
- ↑ Virdis, G.; Dessole, M.; Dessole, S.; Ambrosini, G.; Cosmi, E.; Cherchil, P. L.; Capobianco, G. (2016-01-01). "Holt Oram syndrome: a case report and review of the literature". Clinical and Experimental Obstetrics & Gynecology. 43 (1): 137–139. ISSN 0390-6663. PMID 27048037.
- ↑ Holt, Mary; Oram, Samuel (1960-04-01). "Familial Heart Disease with Skeletal Malformations". British Heart Journal. 22 (2): 236–242. doi:10.1136/hrt.22.2.236. ISSN 0007-0769. PMC 1017650. PMID 14402857.
Further reading
- GeneReview/NIH/UW entry on Holt-Oram Syndrome Archived 2020-12-01 at the Wayback Machine
External links
| Classification | |
|---|---|
| External resources |
|