Acrocallosal syndrome
| Acrocallosal syndrome | |
|---|---|
 | |
| Polydactyly and hallux duplication in a 1 day old infant due to acrocallosal syndrome. | |
Acrocallosal syndrome (also known as ACLS) is a rare autosomal recessive syndrome characterized by corpus callosum agenesis, polydactyly, multiple dysmorphic features, motor and intellectual disabilities, and other symptoms.[1] The syndrome was first described by Albert Schinzel in 1979.[2]
It is associated with GLI3.[3]
Signs and symptoms
Acrocallosal syndrome (ACLS, ACS, Schinzel-Type, Hallux-duplication) is a rare, heterogeneous[3] autosomal recessive disorder first discovered by Albert Schinzel (1979) in a 3 year-old boy.[2] To inherit ACLS, one gene copy from each parent must contain a mutation somewhere in the KIF7 gene and be passed on to the child. Characteristics of this syndrome include absence or poor development of the area connecting the left and right parts of the brain, an abnormally large head, increased distance between facial features (eyes), poor motor skills, mental retardation, extra fingers and toes, many facial deformities,[3] and cleft palate.
Acrocallosal syndrome is considered a rare disorder and is placed on the NIH Office of Rare Diseases (fewer than 200,000 cases) rare disease list. Lifespan may range from stillbirth to normal expectancy depending on pregnancy complications and severity of the disorder.[2] In mild cases, the subjects have been shown to live relatively normal lives, but with developmental delays.[2]
Pathology

Acrocallosal syndrome (ACLS, ACS, Schinzel-Type) is a rare, heterogeneous, autosomal recessive disorder.[3] The heterogeneity of this condition refers to the multiple genes that may function to contribute to varying degrees of this syndrome[3] and is often linked to consanguinity.[2] Characteristics of this syndrome include agenesis of the corpus, macrocephaly, hypertelorism, polydactyly, mental and motor retardation,[2] craniofacial dysmorphism, hallux duplication,[3] and sometimes palatal clefting. It has also been reported that there are many similar signs and symptoms between ACLS, Greig cephalopolysyndactyly, and hydrolethalus syndrome (HLS), although there is little evidence to support common genetic causation at this point.[3]
Mechanism
Recent studies by, have indicated that the mutations responsible for ACLS are located in the KIF7 protein. KIF7 is a 1343 amino acid protein with a kinesin motor, coiled coil, and Gli-binding domains. Its functions are largely associated with ciliary motor function,[3] and is a key factor in the Sonic Hedgehog (Shh) transduction pathways that are crucial during embryogenesis . Therefore, mutations in Hedgehog signaling components (KIF7) may lead to cilia based pathologies and ultimately defects in the brain and other areas associated with ACLS and related disorders . More specifically, mutations observed by, in the GLI3 gene have yielded similar characteristics to ACLS, but further evidence is needed since the corpus callosum is usually present in these cases. Other studies have also indicated that KIF7 interacts with Gli transcription factors, so mutations in the KIF7 gene may be upstream effectors of GLI3. Further understanding about the functionality of ACLS and its underlying mechanisms opens new doors to pharmacological manipulation and other forms of molecular therapy.[4]
Diagnosis
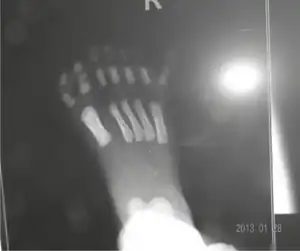
The diagnosis of this condition is done by meeting some of the following criteria (molecular testing can confirm said diagnosis):[5]
- Partial absence of CC
- Minor craniofacial anomalies
- Severe global developmental delay
- Polydactyly
Differential diagnosis
The DDX is based on the following:[5]
- Smith-Lemli-Opitz
- Greig cephalopolysyndactyly
- Meckel-Gruber
Treatment
In terms of the management pf this condition surgery can assist the polydactyly clinical presentation. Management is also supportive with special education programs.[5]
References
- ↑ Online Mendelian Inheritance in Man (OMIM): Acrocallosal syndrome; ACLS - 200990
- 1 2 3 4 5 6 Schinzel, Albert (May 1979). "Postaxial polydactyly, hallux duplication, absence of the corpus callosum, macroencephaly and severe mental retardation: a new syndrome?". Helvetica Paediatrica Acta. 34 (2): 141–6. PMID 457430.
- 1 2 3 4 5 6 7 8 Elson E, Perveen R, Donnai D, Wall S, Black GC (November 2002). "De novo GLI3 mutation in acrocallosal syndrome: broadening the phenotypic spectrum of GLI3 defects and overlap with murine models". J. Med. Genet. 39 (11): 804–6. doi:10.1136/jmg.39.11.804. PMC 1735022. PMID 12414818.
- ↑ Putoux, Audrey; Thomas, Sophie; Coene, Karlien L M; Davis, Erica E; Alanay, Yasemin; Ogur, Gönül; Uz, Elif; Buzas, Daniela; Gomes, Céline (2013-06-06). "KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes". Nature Genetics. 43 (6): 601–606. doi:10.1038/ng.826. ISSN 1061-4036. PMC 3674836. PMID 21552264.
- 1 2 3 RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Acrocallosal syndrome". www.orpha.net. Archived from the original on 30 July 2017. Retrieved 21 August 2021.
- Koenig R.; Bach A.; Ulrike W.; Grzeschik K-H; Fuchs S. (2002). "Spectrum of the acrocallosal syndrome". American Journal of Medical Genetics. 108 (1): 7–11. doi:10.1002/ajmg.10236. PMID 11857542.
- Walsh D.; Shalev S.; Simpson M.; Morgan N.; Gelman-Kohan Z.; Chemke J.; Trembath R.; Maher E. (2013). "Acrocallosal syndrome: Identification of a novel KIF7 mutation and evidence for oligogenic inheritance". European Journal of Medical Genetics. 56 (1): 39–42. doi:10.1016/j.ejmg.2012.10.004. PMID 23142271.
- Putoux A.; Nampoothiri S.; Laurent N.; Cormier-Daire V.; Beales P.; et al. (2012). "Novel KIF7 mutations extend the phenotypic spectrum of acrocallosal sndrome". Journal of Medical Genetics. 49 (11): 713–720. doi:10.1136/jmedgenet-2012-101016. PMID 23125460. S2CID 21874554.
- Putoux A.; Thomas S.; Coene K. L. M.; Davis E. E.; Alanay Y.; Ogur G.; et al. (2011). "KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes". Nature Genetics. 43 (6): 601–606. doi:10.1038/ng.826. PMC 3674836. PMID 21552264.
- Elson E.; Perveen R.; Donnai D.; Wall S.; Black G.C.M. (2002). "De novo GLI3 mutation in acrocallosal syndrome:broadening the phenotypic spectrum of GLI3 defects and overlap with murine models". Journal of Medical Genetics. 39 (11): 804–806. doi:10.1136/jmg.39.11.804. PMC 1735022. PMID 12414818.
- Klejnot M.; Kozielski F. (2011). "Structural insights into human Kif7, a kinesin involved in Hedgehog signaling". Acta Crystallographica Section D. 68 (2): 154–159. doi:10.1107/S0907444911053042. PMC 3266853. PMID 22281744.
- NIH Office of Rare Diseases Research. (n.d.). Acrocallosal syndrome, Schinzel type. Bethesda, MD. Retrieved on 2/4/14 from: http://rarediseases.info.nih.gov/gard/5721/acrocallosal-syndrome-schinzel-type/resources/1 Archived 2016-03-16 at the Wayback Machine
External links
| Classification | |
|---|---|
| External resources |
|
- Acrocallosal syndrome, Schinzel type at NIH's Office of Rare Diseases
symptoms of vibes