Kabuki syndrome
| Kabuki syndrome | |
|---|---|
| Other names: Niikawa–Kuroki syndrome | |
 | |
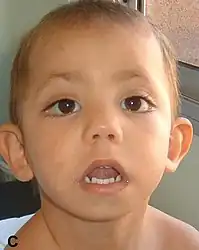
| A child with kabuki syndrome displaying the “scrunchy face” | |
| Symptoms | Vary widely among patients but may include: Long eyelashes, depressed nasal tip, atypical fingerprints, ear deformity (macrotia or microtia), hypotonia, joint hyperflexibility, ptosis, blue sclera, cafe au lait spot, GU anomalies (e.g. hypospadias or horseshoe kidney), gi anomalies (e.g. anal atresia or intestinal malformation), hearing loss, immune deficiencies (e.g. hypogammaglobinemia), feeding difficulty (infants), obesity (adulthood), short stature, poor sleep, hyperinsulinemia (hypoglycemia), epilepsy, cardiac defects (e.g. coarctation of the aorta), vertebral anamolies (e.g. butterfly vertebrae), sparse lateral eyelash, finger anomaly (e.g. short 5th finger), cleft palate, dental issues, precocious puberty, scoliosis, hip dysplasia |
| Usual onset | Conception |
| Types | Type 1 (KMT2D), type 2 (KDM6A); other rare mutations unrecognized for now |
| Causes | Loss-of-function mutations in KMT2D or KDM6A genes |
| Diagnostic method | Clinical findings; genetic testing |
| Frequency | 1 in 32,000 births |
Kabuki syndrome (also previously known as Kabuki-makeup syndrome (KMS) or Niikawa–Kuroki syndrome) is a congenital disorder of genetic origin.[1][2] It affects multiple parts of the body, with varying symptoms and severity, although the most common is the characteristic facial appearance.[3]
It is quite rare, affecting roughly one in 32,000 births.[4] It was first identified and described in 1981 by two Japanese groups, led by scientists Norio Niikawa and Yoshikazu Kuroki.[5] It is named Kabuki syndrome because of the facial resemblance of affected individuals to stage makeup used in kabuki, a Japanese traditional theatrical form.[4]
Signs and symptoms
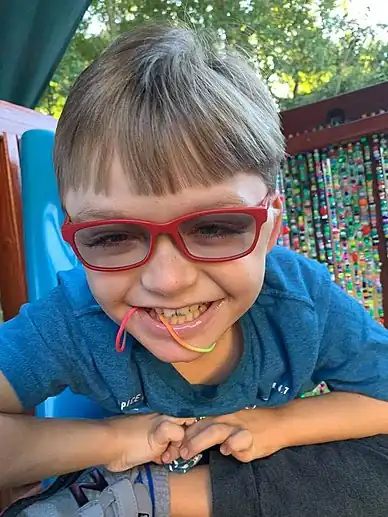
Specific symptoms for Kabuki syndrome vary, with large differences between affected individuals.[3] Most people with Kabuki syndrome have distinctive facial features that include arched eyebrows, long eyelashes, elongated eyelids with lower lids that turn out, prominent ears, a flat tip of the nose and a downward slant to the mouth.[3][4]
Overlapping phenotypic features for patients between KDM6A and KDM6B variations include prominent ears, abnormal dentition, congenital heart disease, feeding difficulties, cryptorchidism, joint hyper-mobility, developmental delay, hypotonia, and behavioral difficulties.
Other common symptoms are skeletal abnormalities, short stature, heart defects, feeding difficulties and a failure to thrive, vision and hearing difficulties, weak muscle tone (hypotonia), small head size (microcephaly), and frequent infections.[3]
Mild to moderate intellectual disability and mild to severe developmental delay are often associated with Kabuki syndrome.[3][4][6] Infants and young children often experience difficulties relating to hypotonia, feeding issues/failure to thrive, infections, surgical repair of heart and palate defects and developmental delays.
Young children with Kabuki syndrome benefit from early intervention services. School age children tend to have fewer medical issues requiring hospitalization, though frequent infections, hearing loss and feeding issues occur. In addition, intellectual impairment, difficulty with visuospatial tasks and maintaining attention usually require an IEP (individualized education plan) if the child attends public school. Older children and adults report difficulties with anxiety. Endocrine abnormalities and immune system abnormalities such as ITP (idiopathic thrombocytopenia) and CVID (common variable immune deficiency) are medical issues that tend to present in older children, adolescents and adults.[7]
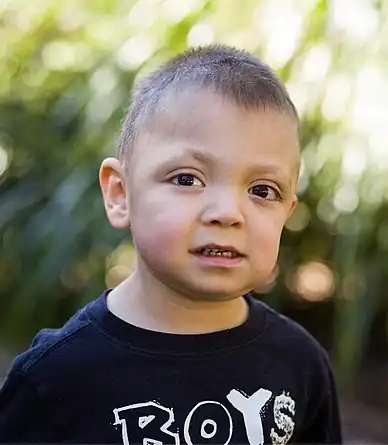
 Child displaying typical facial phenotype of Kabuki syndrome
Child displaying typical facial phenotype of Kabuki syndrome Individual with Kabuki Syndrome from infancy to adulthood
Individual with Kabuki Syndrome from infancy to adulthood
Causes
Kabuki syndrome is one of the Mendelian disorders of epigenetic machinery.[8] Most cases of Kabuki syndrome occur de novo, that is, the parents are unaffected and the gene was mutated early in embryological development. It is classified as a Mendelian disorder because individuals who have a de novo mutation in a specific gene pass the mutation to offspring according to the laws of Mendelian inheritance. There are two known genes that cause Kabuki syndrome: KMT2D and KDM6A.[9] However, about 30% of cases have no identifiable causative mutation.[4]
It is estimated that between 55-80% of cases of Kabuki syndrome are caused by mutations in the KMT2D gene, formerly known as the MLL2 gene.[4] This gene is located on chromosome 12.[4][9] A mutation in the KMT2D gene results in a non functional lysine (K)-specific methyltransferase 2D enzyme[4] and demonstrates an autosomal dominant pattern of inheritance. Another 2-6% of cases are related to mutations in the KDM6A gene, located on the X chromosome.[4] This mutation produces a nonfunctional lysine (K)-specific demethylase 6A enzyme and demonstrates an X-linked dominant pattern of inheritance.
Pathophysiology
The KMT2D and KDM6A genes belong to a family of genes called chromatin-modifying enzymes. Specifically, these genes code for a histone methyltransferase (KMT2D) and a histone demethylase (KDM6A), and play a part in the regulation of gene expression.[10] Under normal circumstances, these enzymes transfer methyl groups on and off histones to regulate genes via epigenetic pathways. When the genes that encode these enzymes are mutated, epigenetic activation of certain developmental genes is impaired and developmental abnormalities occur, leading to the characteristics of Kabuki syndrome patients.[10] The specific developmental genes that are affected by the impaired epigenetic mechanisms in Kabuki syndrome are not yet fully known.[10]
There are hundreds of different mutations that have been identified in Kabuki syndrome patients. Most of these mutations are in the KMT2D gene and involve a change in amino acid sequence that creates a shortened and nonfunctional chromatin-modifying enzyme.[11]
Diagnosis
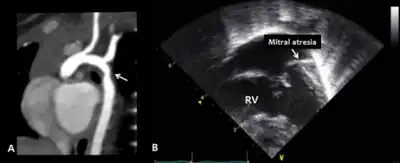
A consensus on clinical diagnostic criteria for Kabuki syndrome (KS) was defined in December 2018 by an international group of experts.[12] The authors propose that a definitive diagnosis can be made in an individual of any age with a history of infantile hypotonia, developmental delay and/or intellectual disability, and one or both of the following major criteria: (1) a pathogenic or likely pathogenic variant in KMT2D or KDM6A; and (2) typical dysmorphic features (defined below) at some point of life. Typical dysmorphic features include long palpebral fissures with eversion of the lateral third of the lower eyelid and two or more of the following: (1) arched and broad eyebrows with the lateral third displaying notching or sparseness; (2) short columella with depressed nasal tip; (3) large, prominent or cupped ears; and (4) persistent fingertip pads. Further criteria for a probable and possible diagnosis, including a table of suggestive clinical features, were included in the publication.[12]
The original description of Kabuki syndrome by Niikawa et al.[13] defined five cardinal manifestations, although some of these “cardinal manifestations” may or may not be present in a patient with Kabuki syndrome.
- Typical facial features: Elongated palpebral fissures with eversion of the lateral third of the lower eyelid; arched and broad eyebrows with the lateral third displaying sparseness or notching; short columella with depressed nasal tip; large, prominent, or cupped ears[13][14][15][16][17]
- Skeletal anomalies: Spinal column abnormalities, including sagittal cleft vertebrae, butterfly vertebrae, narrow intervertebral disc space, and/or scoliosis, Brachydactyly V Brachymesophalangy Clinodactyly of fifth digits
- Dermatoglyphic abnormalities: persistence of fetal fingertip pads
- Mild to moderate intellectual disability
- Postnatal growth deficiency
Kabuki syndrome is diagnosed clinically (through identifying symptoms, physical exams, and lab results), most commonly by a geneticist. Alternatively, it may be discovered using genetic testing (whole exome or whole genome sequencing).[3]
Diagnosis can be difficult given the large spectrum of disease. The fact that some patients do not carry one of the two known mutations or can carry multiple mutations complicates the diagnosis further.
Screening
Due to its rarity, Kabuki syndrome is not screened for in routine prenatal testing including blood tests, chorionic villus sampling (CVS), or amniocentesis. Although not routine for the general population, if Kabuki syndrome is a specific concern (i.e. expectant mother who has been diagnosed with Kabuki syndrome or sibling with KS), it is possible to test for one of the specific mutations.[4] This prenatal testing does require a CVS or amniocentesis. However Kabuki syndrome is usually not inherited and therefore most cases do not have a positive family history.[3][4] Kabuki syndrome can have positive screening tests, such as cystic hygroma seen on nuchal translucency ultrasound screening, although these findings are non-specific and have a wide differential diagnosis.[18][19]
Management
Newly diagnosed patients with Kabuki syndrome will often undergo tests that are aimed at detecting common abnormalities associated with the syndrome. They include an echocardiogram (ultrasound of the heart) for detection of structural heart defects, kidney ultrasound for detection of structural renal abnormalities, immunoglobulin levels, pneumococcal titers and a hearing screening test. Further evaluation and testing by specialists may be indicated in addition to cardiology, nephrology, allergy/immunology, audiology-mentioned above. This may include orthopedics (such as hip dysplasia), pulmonary (sleep study to rule out obstructive sleep apnea due to hypotonia), ophthalmology evaluation (vision screen), ENT evaluation (hearing evaluation), Neurology evaluation (i.e. if seizures present), Hematology evaluation (if bleeding disorder), GI evaluation (if gi abnormalities), or others as needed.
There is no specific treatment for Kabuki syndrome. Treatment plans are customized to address the symptoms the individual is experiencing.[10] For example, someone experiencing seizures will be treated with the standard anti-epilepsy therapies.[10] Additionally, patients with Kabuki syndrome are routinely evaluated and monitored to address problems that may develop, such as vision or hearing problems, or cognitive difficulties.[10] If congenital heart disease is present, prophylactic antibiotics may be recommended before any procedures such as dental work that might cause infection.[10]
Prognosis
Life expectancy is not shortened in most cases of Kabuki syndrome. Some patients have coexisting conditions which may shorten life expectancy, such as hypoplastic left heart syndrome or kidney dysfunction. It is important that patients with cardiac, renal, or immunologic issues are identified and well-managed.[3]
Epidemiology
Kabuki syndrome occurs about once in every 32,000 births.[4][13] The disease appears to affect all population groups equally, with no differences based on sex, race, or environment.[20]
Research
Research on Kabuki syndrome is extremely limited due to its low incidence.[4] Despite this, several groups around the world are studying Kabuki syndrome. In the United States, these include the Epigenetics and Chromatin Clinic at Johns Hopkins University (led by Dr. Hans Bjornsson), The Roya Kabuki Program at Boston Children's Hospital, Dr. Mark Hannibal at the University of Michigan, groups at University of Colorado, University of Utah, University of South Florida and others.[21][22]
History
In 1969, Norio Niikawa MD, a geneticist in Japan was treating a child patient presenting with unique facial characteristics and various health problems. Never having seen this constellation of symptoms before, Dr Niikawa wondered if he was faced with an undiagnosed condition, a disorder with a genetic basis. Over the next several years, this physician treated several other patients with the same symptoms in his outpatient genetics clinic, furthering support for a disorder never before diagnosed.[23]
In 1979, Dr Niikawa presented his findings and hypothesis at the first Japan Dysmorphology Conference. A fellow physician at this conference, Yoshikazu Kuroki, recognised the symptoms, and realised that he had also seen several paediatric patients with this presentation; he presented two of his own cases at the second annual conference the following year. In 1981, the two doctors separately submitted articles on this new diagnosis to the Journal of Pediatrics.[5][23][24]
Dr Niikawa coined the term ‘Kabuki syndrome’ (also known as Kabuki make-up syndrome or Niikawa–Kuroki syndrome) as a reference to traditional Japanese theatre which he respected greatly. Many of the children presenting with this diagnosis had unusual, elongated lower eyelids, and this feature was reminiscent of the theatrical make-up worn by actors in Kabuki theatre.[25][26]
As reported by Dr. Niikawa "The name, “Kabuki make-up”, of the syndrome was given by myself, because the facial appearance of patients, especially eversion of their lower eyelids, is reminiscent of the makeup of actors in Kabuki, the traditional form of Japanese theater. Kabuki was founded early in the 17th century in Japan and over the next 300 years developed into a sophisticated form of theater. Kabuki actors usually apply traditional makeup to strengthen their eyes, especially in a hero play, and they are very proud of their performing art."[27]
The individual kanji, from left to right, mean sing (歌), dance (舞), and skill (伎). Kabuki is therefore sometimes translated as "the art of singing and dancing".
Gallery




References
- ↑ "Kabuki Syndrome Gene Identified". National Institutes of Health (NIH). 2015-05-20. Archived from the original on 2015-10-05. Retrieved 2017-10-26.
- ↑ Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. (September 2010). "Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome". Nature Genetics. 42 (9): 790–3. doi:10.1038/ng.646. PMC 2930028. PMID 20711175.
- 1 2 3 4 5 6 7 8 "Kabuki syndrome". Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. Archived from the original on 2018-03-16. Retrieved 2018-04-01.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 "Kabuki syndrome". Genetics Home Reference, U.S. National Library of Medicine. Archived from the original on 21 April 2018. Retrieved 15 April 2018.
- 1 2 Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I (October 1981). "A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation". The Journal of Pediatrics. 99 (4): 570–3. doi:10.1016/S0022-3476(81)80256-9. PMID 7277097.
- ↑ Vaux KK, Jones KL, Jones MC, Schelley S, Hudgins L (January 2005). "Developmental outcome in Kabuki syndrome". American Journal of Medical Genetics. Part A. 132A (3): 263–4. doi:10.1002/ajmg.a.30338. PMID 15523636. S2CID 32831210.
- ↑ Adam, Margaret P.; Hudgins, Louanne; Hannibal, Mark (2019-10-21). Kabuki Syndrome. University of Washington, Seattle. PMID 21882399. Archived from the original on 2021-10-20. Retrieved 2021-10-15.
- ↑ Bjornsson HT (October 2015). "The Mendelian disorders of the epigenetic machinery". Genome Research. 25 (10): 1473–81. doi:10.1101/gr.190629.115. PMC 4579332. PMID 26430157.
- 1 2 Cheon CK, Sohn YB, Ko JM, Lee YJ, Song JS, Moon JW, Yang BK, Ha IS, Bae EJ, Jin HS, Jeong SY (June 2014). "Identification of KMT2D and KDM6A mutations by exome sequencing in Korean patients with Kabuki syndrome". Journal of Human Genetics. 59 (6): 321–5. doi:10.1038/jhg.2014.25. PMID 24739679. S2CID 24533876.
- 1 2 3 4 5 6 7 Adam MP, Hudgins L, Hannibal M (16 May 2013). Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). "Kabuki Syndrome". GeneReviews [Internet]. PMID 21882399. Archived from the original on 20 October 2021. Retrieved 15 October 2021.
- ↑ Bögershausen N, Wollnik B (March 2013). "Unmasking Kabuki syndrome". Clinical Genetics. 83 (3): 201–11. doi:10.1111/cge.12051. PMID 23131014. S2CID 204999137.
- 1 2 Adam, Margaret P.; Banka, Siddharth; Bjornsson, Hans T.; Bodamer, Olaf; Chudley, Albert E.; Harris, Jaqueline; Kawame, Hiroshi; Lanpher, Brendan C.; Lindsley, Andrew W. (2018-12-04). "Kabuki syndrome: international consensus diagnostic criteria". Journal of Medical Genetics. 56 (2): 89–95. doi:10.1136/jmedgenet-2018-105625. ISSN 1468-6244. PMID 30514738. S2CID 54484060.
- 1 2 3 Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T (October 1981). "Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency". The Journal of Pediatrics. 99 (4): 565–9. doi:10.1016/S0022-3476(81)80255-7. PMID 7277096.
- ↑ Wilson GN (September 1998). "Thirteen cases of Niikawa-Kuroki syndrome: report and review with emphasis on medical complications and preventive management". American Journal of Medical Genetics. 79 (2): 112–20. doi:10.1002/(SICI)1096-8628(19980901)79:2<112::AID-AJMG7>3.0.CO;2-S. PMID 9741469.
- ↑ Hannibal MC, Buckingham KJ, Ng SB, Ming JE, Beck AE, McMillin MJ, et al. (July 2011). "Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome". American Journal of Medical Genetics. Part A. 155A (7): 1511–6. doi:10.1002/ajmg.a.34074. PMC 3121928. PMID 21671394.
- ↑ Schrander-Stumpel CT, Spruyt L, Curfs LM, Defloor T, Schrander JJ (January 2005). "Kabuki syndrome: Clinical data in 20 patients, literature review, and further guidelines for preventive management". American Journal of Medical Genetics. Part A. 132A (3): 234–43. doi:10.1002/ajmg.a.30331. PMID 15690368. S2CID 42186309.
- ↑ Armstrong L, Abd El Moneim A, Aleck K, Aughton DJ, Baumann C, Braddock SR, et al. (January 2005). "Further delineation of Kabuki syndrome in 48 well-defined new individuals". American Journal of Medical Genetics. Part A. 132A (3): 265–72. doi:10.1002/ajmg.a.30340. PMID 15690370. S2CID 27626946.
- ↑ Long A, Sinkovskaya ES, Edmondson AC, Zackai E, Schrier Vergano SA (December 2016). "Kabuki syndrome as a cause of non-immune fetal hydrops/ascites". American Journal of Medical Genetics. Part A. 170 (12): 3333–3337. doi:10.1002/ajmg.a.37956. PMID 27568880. S2CID 26914645.
- ↑ Lajeunesse C, Stadler A, Trombert B, Varlet MN, Patural H, Prieur F, Chêne G (June 2014). "[First-trimester cystic hygroma: prenatal diagnosis and fetal outcome]". Journal de Gynécologie, Obstétrique et Biologie de la Reproduction. 43 (6): 455–62. doi:10.1016/j.jgyn.2013.04.005. PMID 23747217.
- ↑ "Kabuki Syndrome - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Archived from the original on 2018-02-07. Retrieved 2018-04-20.
- ↑ "Kabuki Syndrome | Boston Children's Hospital". www.childrenshospital.org. Archived from the original on 2018-04-24. Retrieved 2018-04-24.
- ↑ Johns Hopkins Medicine (2015-10-07), #TomorrowsDiscoveries: Intellectual Disability Treatments — Dr. Hans Bjornsson, archived from the original on 2021-10-20, retrieved 2018-04-24
- 1 2 Matsumoto, Naomichi; Niikawa, Norio (2003-01-30). "Kabuki make-up syndrome: A review". American Journal of Medical Genetics. 117C (1): 57–65. doi:10.1002/ajmg.c.10020. ISSN 0148-7299. PMID 12561059. S2CID 39599820.
- ↑ Niikawa, Norio; Matsuura, Nobuo; Fukushima, Yoshimitsu; Ohsawa, Tadashi; Kajii, Tadashi (October 1981). "Kabuki make-up syndrome: A syndrome of mentalretardation, unusual facies, large and protruding ears, and postnatal growth deficiency". The Journal of Pediatrics. 99 (4): 565–569. doi:10.1016/s0022-3476(81)80255-7. ISSN 0022-3476. PMID 7277096.
- ↑ Schrander-Stumpel, Constance Th.R.M.; Spruyt, Liesbeth; Curfs, Leopold M.G.; Defloor, Truus; Schrander, Jaap J.P. (2004-12-03). "Kabuki syndrome: Clinical data in 20 patients, literature review, and further guidelines for preventive management". American Journal of Medical Genetics Part A. 132A (3): 234–243. doi:10.1002/ajmg.a.30331. ISSN 1552-4825. PMID 15690368. S2CID 42186309.
- ↑ Kasdon, Bethany D; Fox, Judith E (September 2012). "Kabuki syndrome: diagnostic and treatment considerations". Mental Health in Family Medicine. 9 (3): 171–179. ISSN 1756-834X. PMC 3622909. PMID 23997823.
- ↑ "Niikawa Presentation" (PDF). Archived (PDF) from the original on 2018-07-18. Retrieved 2021-10-15.
External links
| Website | |
|---|---|
| Patient Advocacy | All Things Kabuki Archived 2021-03-08 at the Wayback Machine |
| Supporting Aussie Kids with Kabuki Syndrome (SAKKS) Archived 2021-05-07 at the Wayback Machine | |
| Association Syndrome Kabuki Archived 2021-05-07 at the Wayback Machine | |
| Research | Kabuki Syndrome Foundation Archived 2021-05-26 at the Wayback Machine |
| Roya Kabuki Program Archived 2021-05-06 at the Wayback Machine | |
| Conferences/Gatherings | Texas Kabuki Gathering Archived 2021-05-07 at the Wayback Machine |
| Classification | |
|---|---|
| External resources |
|