Stargardt disease
| Stargardt disease | |
|---|---|
| Other names: Stargardt macular dystrophy & degeneration, juvenile macular degeneration, fundus flavimaculatus | |
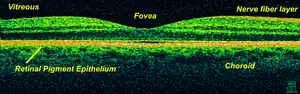 | |
| Optical coherence tomography is used for diagnosis of Stargardt's disease. | |
| Specialty | Ophthalmology |
| Symptoms | Blurred vision |
| Usual onset | Childhood |
| Duration | Lifelong |
| Causes | Genetic |
| Diagnostic method | Slit-lamp |
| Treatment | None |
Stargardt disease is the most common inherited single-gene retinal disease.[1] It usually has an autosomal recessive inheritance caused by mutations in the ABCA4 gene. Rarely it has an autosomal dominant inheritance due to defects with ELOVL4 or PROM1 genes. It is characterised by macular degeneration that begins in childhood, adolescence or adulthood, resulting in progressive loss of vision.[2]
Signs and symptoms
Presentation usually occurs in childhood or adolescence, though there is no upper age limit for presentation and late onset is possible. The main symptom is loss of visual acuity, uncorrectable with glasses. This manifests as the lack of the ability to see fine details when reading or viewing distant objects. Symptoms typically develop before age 20 (median age of onset: ~17 years old),[3] and include: wavy vision, blind spots, blurriness, loss of depth perception, sensitivity to glare, impaired colour vision,[3] and difficulty adapting to dim lighting (delayed dark adaptation). There is a wide variation between individuals in the symptoms experienced as well as the rate of deterioration in vision. Vision loss can be attributed to buildup of byproducts of vitamin A in photoreceptor cells and Peripheral vision is usually less affected than fine, central (foveal) vision.
Genetics
Historically from Stargardt’s first description of his eponymous disease until recently, the diagnosis was based on looking at the phenotype using examination and investigation of the eye. Since the advent of genetic testing, the picture has become more complex. What was thought to be one disease is, in fact, probably at least three different diseases, each related to a different genetic change. Therefore it is currently a little confusing to define what Stargardt's disease is. It is certainly caused by defects in the ABCA4 gene, but whether changes to other genes such as PROM1 or ELOVL4, or missense mutations play a role remains to be seen.
The carrier frequency in the general population of ABCA4 alleles is 5 to 10%.[4] Different combinations of ABCA4 genes will result in widely different age of onset and retinal pathology. The severity of the disease is inversely proportional to ABCA4 function and it is thought that ABCA4 related disease has a role to play in other diseases such as retinitis pigmentosa, cone-rod dystrophies and age-related macular degeneration (AMD).[5]
- STGD1: By far the most common form of Stargardt disease is the recessive form caused by mutations in the ABCA4 gene.[6]
- STGD4: A rare dominant defect in the PROM1 gene.[7][5]
- STGD3: A rare dominant form of Stargardt disease caused by mutations in the ELOVL4 gene.
- Late-onset Stargardt disease is associated with missense mutations outside known functional domains of ABCA4.[5]
Pathophysiology
In STGD1, the genetic defect causes malfunction of the ATP-binding cassette transporter (ABCA4) protein of the visual phototransduction cycle. Defective ABCA4 leads to improper shuttling of vitamin A throughout the retina, and accelerated formation of toxic vitamin A dimers (also known as bisretinoids), and associated degradation byproducts. Vitamin A dimers and other byproducts are widely accepted as the cause of STGD1. As such, slowing the formation of vitamin A dimers might lead to a treatment for Stargardt. When vitamin A dimers and byproducts damage the retinal cells, fluorescent granules called lipofuscin in the retinal pigmented epithelium of the retina[8] appear, as a reflecting such damage.
In STGD4, a butterfly pattern of dystrophy is caused by mutations in a gene that encodes a membrane bound protein that is involved in the elongation of very long chain fatty acids (ELOVL4)[9]
Diagnosis
Diagnosis is firstly clinical through history and examination usually with a Slit-lamp. If characteristic features are found the investigations undertaken will depend on locally available equipment and may include Scanning laser ophthalmoscopy which highlights areas of autofluorescence which are associated with retinal pathology. Spectral-domain optical coherence tomography, electroretinography and microperimetry are also useful for diagnostic and prognostic purposes. Fluorescein angiography is used less often than in the past. These investigations may be followed by genetic testing in order to avoid misdiagnosis. Other diseases may have overlapping phenotypic features with Stargardt Disease and the disease itself has multiple variants. In one study 35% of patients diagnosed with Stargardt Disease through physical ophthalmic examination were found to be misdiagnosed when subsequent genetic testing was done.[10]
Genetic testing can be utilized to ensure an proper diagnosis for which the correct treatment can be applied.
Treatment
At present there is no treatment for Stargardt Disease. However, ophthalmologists recommend measures that could slow the rate of progression. There are no prospective clinical trials to support these recommendations, but they are based on scientific understanding of the mechanisms underlying the disease pathology. There are three strategies doctors recommend for potential harm reduction: reducing retinal exposure to damaging ultra violet light, avoiding excess Vitamin A with the hope of lowering lipofuscin accumulation and maintaining good general health and diet.
Ultra-violet light has more energy and is more damaging colour than visible light. In an effort to mitigate this, some ophthalmologists may recommend that the patient wears a broad-brimmed hat or sunglasses when they are outdoors.[11] Sometimes, doctors also instruct their patients to wear yellow-tinted glasses (which filter out blue light) when indoors and in artificial light or in front of a digital screen.
Certain foods, especially carrots, are rich in vitamin A, but the amount from food is not harmful.[11] Foods with a high vitamin A content are often yellow or orange in color, such as squash, pumpkin, and sweet potato, but some, such as liver, are not. There are supplements on the market with more than a daily allowance of vitamin A that should be avoided, but each individual should discuss this with their doctor.
Smoking, being overweight, and eating unhealthily may also contribute to more rapid degeneration. On the other hand, the consumption of oily fish, in a diet similar to that which doctors recommend for age related macular degeneration, can be used to slow the progression of the disease.
Advances in technology have brought devices that help Stargardt patients who are losing their vision maintain their independence. Low-vision aids can range from hand lenses to electronic devices and can allow those losing their vision to be able to carry out daily activities. [11] Some patients may even opt for in-person services.
Prognosis
The long-term prognosis for patients with Stargardt disease is widely variable and depends on the age of onset and genetic alleles. The majority of people will progress to legal blindness.[12] Stargardt disease has no impact on general health and life expectancy is normal.[13] Some patients, usually those with the late onset form, can maintain excellent visual acuities for extended periods, and are therefore able to perform tasks such as reading or driving.[9]
Epidemiology
A 2017 prospective epidemiologic study which recruited 81 patients with STGD over 12 months reported an incidence of between 1 and 1.28 per 10 000 individuals. The median age of presentation was 27 years (range 5–64 years), most (90%) were symptomatic, with a median visual acuity of Snellen equivalent 20/66.[14]
History
Karl Stargardt (1875–1927) was a German ophthalmologist born in Berlin. He studied medicine at the University of Kiel, qualifying in 1899. He later became head of the Bonn University’s ophthalmology clinic, followed by a post as chair of ophthalmology at the University of Marburg. In 1909 he described 7 patients with a recessively inherited macular dystrophy, now known as Stargardt’s disease.[15][16][17]
Research
There are early stage clinical trials involving several potential therapeutic areas, gene therapy, stem cell therapy, drug therapy and artificial retinas. In general all are testing the safety and benefits of their respective therapies in phase I or II trials. These studies are designed to evaluate the safety, dose and effectiveness in small number of people in Phase I with Phase II evaluating similar criteria in a larger population but including a greater insight into potential side effects.
Gene therapy is designed to insert a copy of a corrected gene into retinal cells. The hope is to return cell function back to normal and the treatment has the potential to stop disease progression. This therapy will not restore impaired vision back to normal. The research is being undertaken by a partnership between Sanofi and Oxford BioMedica. A Lentiviral vector is used to deliverer normal genes to the eye via a subretinal injection. The therapy is known as SAR422459 and it has been terminated prematurely due to halt in developing the drug product. [18] Kubota Vision published the results of a dose range study of a drug known as Emixustat, with findings that will effect dose selection for their phase III trial set to complete in June 2022. [19]
Stem-cell therapy involves injecting cells with the potential to mature into differentiated and functioning retinal cells. This therapy has the potential stop disease progression and in the long term improve vision. To improve vision this technique will need to replicate the complex multi-layered and neurally anatomy of the retina. There are a number of research groups working with stem cells one of which is Ocata Therapeutics.[20]
Alkeus Pharma is evaluating the potential of deuterated vitamin A as the drug ALK-001. The hope is that the deuterated vitamin A will reduce the build-up of toxic vitamin A metabolites in the retina and therefore slow rate of visual deterioration. To create deuterated vitamin A some of the hydrogen atoms are replaced with the isotope deuterium which has an extra neutron and is therefore twice the standard atomic weight of hydrogen. A Phase II clinical trial is taking place using ALK-001 with an estimated completion date of December 2024.[18][21][22]
MD Stem Cells, a research-physician clinical development company using autologous bone marrow derived stem cells (BMSC), has released results of the Stargardt Disease cohort within their ongoing Stem Cell Ophthalmology Study II (SCOTS2) clinical trial (NCT 03011541).[23] Average visual improvement was 17.96% (95% CI, 16.39 to 19.53%) with 61.8% of eyes improving and 23.5% remaining stable with no adverse events occurring.[24]
Retinal implants are in the early stages of development and their use could be of benefit to many people with Visual impairment though implanting and maintaining an electrical device within the eye that interfaces with the optic nerve presents many challenges. An example of a device is made by Argus retinal prosthesis, the camera is an external device held on spectacles, the camera signal is processed and then fed via wires into the retina to terminate in some electrodes that interface with the optic nerve.[25]
References
- ↑ Clinical Characteristics and Current Therapies for Inherited Retinal Degenerations Jose ́ -Alain Sahel
- ↑ "Stargardt disease : Definition(s) from the Unified Medical Language System ® Diseases Database". diseasesdatabase.com. Retrieved 5 February 2018.
{{cite web}}: CS1 maint: url-status (link) - 1 2 "Stargardt disease/Fundus flavimaculatus". eyewiki.aao.org. Archived from the original on 5 February 2018. Retrieved 5 February 2018.
- ↑ Yatsenko et al. 2001
- 1 2 3 "Stargardt disease/Fundus flavimaculatus – EyeWiki". Archived from the original on 2018-02-05. Retrieved 2021-08-20.
- ↑ "Archive copy". Archived from the original on 2021-03-23. Retrieved 2021-08-20.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ "Archive copy". Archived from the original on 2021-01-30. Retrieved 2021-08-20.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ Adler L, 4th; Boyer, NP; Chen, C; Ablonczy, Z; Crouch, RK; Koutalos, Y (2015). The 11-cis Retinal Origins of Lipofuscin in the Retina. Progress in Molecular Biology and Translational Science. Vol. 134. pp. e1–12. doi:10.1016/bs.pmbts.2015.07.022. ISBN 9780128010594. PMID 26310175.
- 1 2 Deutman, August; Hoyng, Carol; van Lith-Verhoeven, Janneke (2006). "Macular dystrophies". Retina (4 ed.). Elsevier Mosby. pp. 1171–74.
- ↑ Ibanez, Manuel Benjamin; Guimarães, Thales Antonio Cabral; Capasso, Jenina; Bello, Nicholas; Levin, Alex V. (March 2021). "Stargardt misdiagnosis: How ocular genetics helps". American Journal of Medical Genetics Part A. 185 (3): 814–19. doi:10.1002/ajmg.a.62045. ISSN 1552-4825. Archived from the original on 2021-04-28. Retrieved 2021-08-20.
- 1 2 3 "Stargardt Disease | National Eye Institute". www.nei.nih.gov. Archived from the original on 2019-07-25. Retrieved 2021-04-25.
- ↑ Yanoff, Myron; Duker, Jay S. (2008). Ophthalmology (3rd ed.). Edinburgh: Mosby. pp. 560–562. ISBN 978-0323057516.
- ↑ Stargardt Disease Archived 2020-09-18 at the Wayback Machine from The University of Arizona College of Medicine, Department of Ophthalmology and Vision Science. Retrieved Jan 2012
- ↑ The Epidemiology of Stargardt Disease in the United Kingdom, Kurt Spiteri Cornish, FRCOphth, Jason Ho, FRCOphth, Susan Downes, FRCOphth, Neil W. Scott, PhD, James Bainbridge, PhD, Noemi Lois, MD, PhD, 2017, American Academy of Ophthalmology
- ↑ synd/2306 at Who Named It?
- ↑ K. B. Stargardt (1909), "Über familiäre, progressive Degeneration in der Makulagegend des Auges", Albrecht von Graefes Archiv für Ophthalmologie (in German), vol. 71, no. 3, pp. 534–550, doi:10.1007/BF01961301, S2CID 12557316, archived from the original on 2020-02-21, retrieved 2021-08-20
{{citation}}: CS1 maint: unrecognized language (link) - ↑ Stargardt, K. (1909). "Über familiäre, progressive Degeneration in der Maculagegend des Auges". Albrecht von Græfe's Archiv für Ophthalmologie. 71 (3): 534–550. doi:10.1007/BF01961301. S2CID 12557316. Archived from the original on 2020-02-21. Retrieved 2021-08-20.
- 1 2 "Home – ClinicalTrials.gov". clinicaltrials.gov. Archived from the original on 2021-04-24. Retrieved 2021-04-25.
- ↑ Kubota, Ryo; Birch, David G.; Gregory, Jeff K.; Koester, John M. (2020-11-19). "Randomised study evaluating the pharmacodynamics of emixustat hydrochloride in subjects with macular atrophy secondary to Stargardt disease". British Journal of Ophthalmology. doi:10.1136/bjophthalmol-2020-317712. ISSN 0007-1161. PMID 33214244. Archived from the original on 2021-04-28. Retrieved 2021-08-20.
- ↑ Schwartz, SD; Regillo, CD; Lam, BL; Eliott, D; Rosenfeld, PJ; Gregori, NZ; Hubschman, JP; Davis, JL; Heilwell, G; Spirn, M; Maguire, J; Gay, R; Bateman, J; Ostrick, RM; Morris, D; Vincent, M; Anglade, E; Del Priore, LV; Lanza, R (7 February 2015). "Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt's macular dystrophy: follow-up of two open-label phase 1/2 studies". Lancet. 385 (9967): 509–16. doi:10.1016/s0140-6736(14)61376-3. PMID 25458728. S2CID 85799.
- ↑ "Archive copy". Archived from the original on 2021-07-14. Retrieved 2021-08-20.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ "Stargardt disease: The leading cause of juvenile macular degeneration". Alkeus Pharma. Archived from the original on 2020-06-23. Retrieved 2021-08-20.
- ↑ "Archive copy". Archived from the original on 2021-08-29. Retrieved 2021-08-20.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ Weiss JN, Levy S. "Stem Cell Ophthalmology Treatment Study (SCOTS): Bone Marrow-Derived Stem Cells in the Treatment of Stargardt Disease". Medicines 2021, 8(2), 10
- ↑ Chuang, AT; Margo, CE; Greenberg, PB (July 2014). "Retinal implants: a systematic review". The British Journal of Ophthalmology. 98 (7): 852–56. doi:10.1136/bjophthalmol-2013-303708. PMID 24403565. S2CID 25193594.
External links
| Classification | |
|---|---|
| External resources |
|
- NCBI Genetic Testing Registry Archived 2018-12-04 at the Wayback Machine