Vitamin A
 Retinol | |
| Names | |
|---|---|
IUPAC name
| |
| Clinical data | |
| Drug class | vitamin |
| WHO AWaRe | UnlinkedWikibase error: ⧼unlinkedwikibase-error-statements-entity-not-set⧽ |
| Pregnancy category |
|
| Routes of use | by mouth, IM[1] |
| External links | |
| AHFS/Drugs.com | Monograph |
| Legal | |
| License data | |
| Legal status |
|
| Chemical and physical data | |
| Formula | C20H30O |
| Molar mass | 286.4516 g·mol−1 |
| 3D model (JSmol) | |
| Melting point | 62–64 °C (144–147 °F) |
| Boiling point | 137–138 °C (279–280 °F) (10−6 mm Hg) |
SMILES
| |
InChI
| |
Vitamin A is a fat-soluble vitamin and an essential nutrient for humans. It is a group of organic compounds that includes retinol, retinal (also known as retinaldehyde), retinoic acid, and several provitamin A carotenoids (most notably beta-carotene [β-carotene]).[2][3][4][5] Vitamin A has multiple functions: it is essential for embryo development and growth, for maintenance of the immune system, and for vision, where it combines with the protein opsin to form rhodopsin – the light-absorbing molecule necessary for both low-light (scotopic vision) and color vision.[6]
Vitamin A occurs as two principal forms in foods: A) retinol, found in animal-sourced foods, either as retinol or bound to a fatty acid to become a retinyl ester, and B) the carotenoids alpha-carotene, β-carotene, gamma-carotene, and the xanthophyll beta-cryptoxanthin (all of which contain β-ionone rings) that function as provitamin A in herbivore and omnivore animals which possess the enzymes that cleave and convert provitamin carotenoids to retinal and then to retinol.[7] Some carnivore species lack this enzyme. The other carotenoids have no vitamin activity.[5]
Dietary retinol is absorbed from the digestive tract via passive diffusion. Unlike retinol, β-carotene is taken up by enterocytes by the membrane transporter protein scavenger receptor B1 (SCARB1), which is upregulated in times of vitamin A deficiency.[5] Storage of retinol is in lipid droplets in the liver. A high capacity for long-term storage of retinol means that well-nourished humans can go months on a vitamin A- and β-carotene-deficient diet, while maintaining blood levels in the normal range.[3] Only when the liver stores are nearly depleted will signs and symptoms of deficiency show.[3] Retinol is reversibly converted to retinal, then irreversibly to retinoic acid, which activates hundreds of genes.[8]
Vitamin A deficiency is common in developing countries, especially in Sub-Saharan Africa and Southeast Asia. Deficiency can occur at any age but is most common in pre-school-age children and pregnant women, the latter due to a need to transfer retinol to the fetus. Vitamin A deficiency is estimated to affect approximately one-third of children under the age of five around the world, resulting in hundreds of thousands of cases of blindness and deaths from childhood diseases because of immune system failure.[9] Reversible night blindness is an early indicator of low vitamin A status. Plasma retinol is used as a biomarker to confirm vitamin A deficiency. Breast milk retinol can indicate a deficiency in nursing mothers. Neither of these measures indicates the status of liver reserves.[5]
The European Union and various countries have set recommendations for dietary intake, and upper limits for safe intake. Vitamin A toxicity also referred to as hypervitaminosis A, occurs when there is too much vitamin A accumulating in the body. Symptoms may include nervous system effects, liver abnormalities, fatigue, muscle weakness, bone, and skin changes, and others. The adverse effects of both acute and chronic toxicity are reversed after consumption of high dose supplements is stopped.[5]
Definition
Vitamin A is a fat-soluble vitamin, a category that also includes vitamins D, E and K. The vitamin encompasses several chemically related naturally occurring compounds or metabolites, i.e., vitamers, that all contain a β-ionone ring.[3] The primary dietary form is retinol, which may have a fatty acid molecule attached, creating a retinyl ester, when stored in the liver. Retinol – the transport and storage form of vitamin A – is interconvertible with retinal, catalyzed to retinal by retinol dehydrogenases and back to retinol by retinaldehyde reductases.[10]
- retinal + NADPH + H+ ⇌ retinol + NADP+
- retinol + NAD+ ⇌ retinal + NADH + H+
Retinal, (also known as retinaldehyde) can be irreversibly converted to all-trans retinoic acid by the action of retinal dehydrogenase
- retinal + NAD+ + H2O → retinoic acid + NADH + H+
Retinoic acid diffuses into the cell nucleus where it regulates more than 500 genes by binding directly to gene targets via retinoic acid receptors.[5]
In addition to retinol, retinal and retinoic acid, there are plant-, fungi- or bacteria-sourced carotenoids which can be metabolized to retinol, and are thus vitamin A vitamers.[11]
There are also what are referred to as 2nd, 3rd and 4th generation retinoids which are not considered vitamin A vitamers because they cannot be converted to retinol, retinal or all-trans retinoic acid. Some are prescription drugs, oral or topical, for various indications. Examples are etretinate, acitretin, adapalene, bexarotene, tazarotene and trifarotene.[12][13]
Absorption, metabolism and excretion
Retinyl esters from animal-sourced foods (or synthesized for dietary supplements for humans and domesticated animals) are acted upon by retinyl ester hydrolases in the lumen of the small intestine to release free retinol. Retinol enters intestinal absorptive cells by passive diffusion. Absorption efficiency is in the range of 70 to 90%. Humans are at risk for acute or chronic vitamin A toxicity because there are no mechanisms to suppress absorption or excrete the excess in urine.[4] Within the cell, retinol is there bound to retinol binding protein 2 (RBP2). It is then enzymatically reesterified by the action of lecithin retinol acyltransferase and incorporated into chylomicrons that are secreted into the lymphatic system.
Unlike retinol, β-carotene is taken up by enterocytes by the membrane transporter protein scavenger receptor B1 (SCARB1). The protein is upregulated in times of vitamin A deficiency. If vitamin A status is in the normal range, SCARB1 is downregulated, reducing absorption.[5] Also downregulated is the enzyme beta-carotene 15,15'-monooxygenase, coded for by the BCMO1 gene, responsible for symmetrically cleaving β-carotene into retinal.[7] Absorbed β-carotene is either incorporated as such into chylomicrons or first converted to retinal and then retinol, bound to RBP2. After a meal, roughly two-thirds of the chylomicrons are taken up by the liver with the remainder delivered to peripheral tissues. Peripheral tissues also can convert chylomicron β-carotene to retinol.[5][14]
The capacity to store retinol in the liver means that well-nourished humans can go months on a vitamin A deficient diet without manifesting signs and symptoms of deficiency. Two liver cell types are responsible for storage and release: hepatocytes and hepatic stellate cells (HSCs). Hepatocytes take up the lipid-rich chylomicrons, bind retinol to retinol-binding protein 4 (RBP4), and transfer the retinol-RBP4 to HSCs for storage in lipid droplets as retinyl esters. Mobilization reverses the process: retinyl ester hydrolase releases free retinol which is transferred to hepatocytes, bound to RBP4, and put into blood circulation. Other than either after a meal or when consumption of large amounts exceeds liver storage capacity, more than 95% of retinol in circulation is bound to RBP4.[14]
Carnivores
Strict carnivores manage vitamin A differently than omnivores and herbivores. Carnivores are more tolerant of high intakes of retinol because those species have the ability to excrete retinol and retinyl esters in urine. Carnivores also have the ability to store more in the liver, due to a higher ratio of liver HSCs to hepatocytes compared to omnivores and herbivores. For humans, liver content can range from 20 to 30 μg/gram wet weight. Notoriously, polar bear liver is acutely toxic to humans because content has been reported in range of 2,215 to 10,400 μg/g wet weight.[15] As noted, in humans, retinol circulates bound to RBP4. Carnivores maintain R-RBP4 within a tight range while also having retinyl esters in circulation. Bound retinol is delivered to cells while the esters are excreted in the urine.[15] In general, carnivore species are poor converters of ionone-containing carotenoids, and pure carnivores such as felidae (cats) lack the cleaving enzyme entirely. They must have retinol or retinyl esters in their diet.[15]
Herbivores
Herbivores consume ionone-containing carotenoids and convert those to retinal. Some species, including cattle and horses, have measurable amounts of beta-carotene circulating in the blood, and stored in body fat, creating yellow fat cells. Most species have white fat and no beta-carotene in circulation.[15]
Activation and excretion
In the liver and peripheral tissues of humans, retinol is reversibly converted to retinal by the action of alcohol dehydrogenases, which are also responsible for the conversion of ethanol to acetaldehyde. Retinal is irreversibly oxidized to retinoic acid (RA) by the action of aldehyde dehydrogenases. RA regulates the activation or deactivation of genes. The oxidative degradation of RA is induced by RA - its presence triggers its removal, making for a short-acting gene transcription signal. This deactivation is mediated by a cytochrome P450 (CYP) enzyme system, specifically enzymes CYP26A1, CYP26B1 and CYP26C1. CYP26A1 is the predominant form in the human liver; all other human adult tissues contained higher levels of CYP26B1. CYP26C1 is expressed mainly during embryonic development. All three convert retinoic acid into 4-oxo-RA, 4-OH-RA and 18-OH-RA. Glucuronic acid forms water-soluble glucuronide conjugates with the oxidized metabolites, which are then excreted in urine and feces.[8]
Metabolic functions
Other than for vision, the metabolic functions of vitamin A are mediated by all-trans-retinoic acid (RA). The formation of RA from retinal is irreversible. To prevent accumulation of RA it is oxidized and eliminated fairly quickly, i.e., has a short half-life. Three cytochromes catalyze the oxidation of retinoic acid. The genes for Cyp26A1, Cyp26B1 and Cyp26C1 are induced by high levels of RA, providing a self-regulating feedback loop.[16][17]
Vision and eye health
Vitamin A status involves eye health via two separate functions. Retinal is an essential factor in rod cells and cone cells in the retina responding to light exposure by sending nerve signals to the brain. An early sign of vitamin A deficiency is night blindness.[5] Vitamin A in the form of retinoic acid is essential to normal epithelial cell functions. Severe vitamin A deficiency, common in infants and young children in southeast Asia causes xerophthalmia characterized by dryness of the conjunctival epithelium and cornea. Untreated, xerophthalmia progresses to corneal ulceration and blindness.[18]
Vision
The role of vitamin A in the visual cycle is specifically related to the retinal compound. Retinol is converted by the enzyme RPE65 within the retinal pigment epithelium into 11-cis-retinal. Within the eye, 11-cis-retinal is bound to the protein opsin to form rhodopsin in rod cells and iodopsin in cone cells. As light enters the eye, the 11-cis-retinal is isomerized to the all-trans form. The all-trans retinal dissociates from the opsin in a series of steps called photo-bleaching. This isomerization induces a nervous signal along the optic nerve to the visual center of the brain. After separating from opsin, the all-trans retinal is recycled and converted back to the 11-cis-retinal form by a series of enzymatic reactions, which then completes the cycle by binding to opsin to reform rhodopsin in the retina.[5] In addition, some of the all-trans retinal may be converted to all-trans retinol form and then transported with an interphotoreceptor retinol-binding protein to the retinal pigmented epithelial cells. Further esterification into all-trans retinyl esters allow for storage of all-trans-retinol within the pigment epithelial cells to be reused when needed. It is for this reason that a deficiency in vitamin A will inhibit the reformation of rhodopsin, and will lead to one of the first symptoms, night blindness.[5][19][20]
Night blindness
Vitamin A deficiency (VAD) caused night blindness is a reversible difficulty for the eyes to adjust to dim light. It is common in young children who have a diet inadequate in retinol and beta-carotene. A process called dark adaptation typically causes an increase in photopigment amounts in response to low levels of illumination. This increases light sensitivity by up to 100,000 times compared to normal daylight conditions. Significant improvement in night vision takes place within ten minutes, but the process can take up to two hours to reach maximal effect.[6] People expecting to work in a dark environment wore red-tinted goggles or were in a red light environment to not reverse the adaptation because red light does not deplete rhodopsin versus what occurs with yellow or green light.[20]
Xerophthalmia and childhood blindness

Xerophthalmia, caused by a severe vitamin A deficiency, is described by pathologic dryness of the conjunctival epithelium and cornea. The conjunctiva becomes dry, thick, and wrinkled. Indicative is the appearance of Bitot's spots, which are clumps of keratin debris that build up inside the conjunctiva. If untreated, xerophthalmia can lead to dry eye syndrome, corneal ulceration and ultimately to blindness as a result of cornea and retina damage. Although xerophthalmia is an eye-related issue, prevention (and reversal) are functions of retinoic acid having been synthesized from retinal rather than the 11-cis-retinal to rhodopsin cycle.[21]
Throughout southeast Asia, estimates are that more than half of children under the age of six years have subclinical vitamin A deficiency and night blindness, with progression to xerophthalmia being the leading cause of preventable childhood blindness.[21] Estimates are that each year there are 350,000 cases of childhood blindness due to vitamin A deficiency.[18] The causes are vitamin A deficiency during pregnancy, followed by low transfer of vitamin A during lactation and infant/child diets low in vitamin A or beta-carotene.[21][18] The prevalence of pre-school age children who are blind due to vitamin A deficiency is lower than expected from incidence of new cases only because childhood vitamin A deficiency significantly increases all-cause mortality.[18]
According to a 2017 Cochrane review, vitamin A deficiency, using serum retinol less than 0.70 µmol/L as a criteria, is a major public health problem affecting an estimated 190 million children under five years of age in low- and middle-income countries, primarily in Sub-Saharan Africa and Southeast Asia. In lieu of or in combination with food fortification programs, many countries have implemented public health programs in which children are periodically given very large oral doses of synthetic vitamin A, usually retinyl palmitate, as a means of preventing and treating VAD. Doses were 50,000 to 100,000 IU (International units) for children aged 6 to 11 months and 100,000 to 200,000 IU for children aged 12 months to five years, the latter typically every four to six months. In addition to a 24% reduction in all-cause mortality, eye-related results were reported. Prevalence of Bitot's spots at follow-up were reduced by 58%, night blindness by 68%, xerophthalmia by 69%.[22]
Gene regulation
RA regulates gene transcription by binding to nuclear receptors known as retinoic acid receptors (RARs; RARα, RARβ, RARγ) which are bound to DNA as heterodimers with retinoid "X" receptors (RXRs; RXRα, RXRβ, RXRγ). RARs and RXRs must dimerize before they can bind to the DNA. Expression of more than 500 genes is responsive to retinoic acid.[5] The process is that RAR-RXR heterodimers recognize retinoic acid response elements on DNA.[23] The receptors undergo a conformational change that causes co-repressors to dissociate from the receptors. Coactivators can then bind to the receptor complex, which may help to loosen the chromatin structure from the histones or may interact with the transcriptional machinery.[24] This response upregulates or downregulates the expression of target genes, including the genes that encode for the receptors themselves.[19] To prevent excess accumulation of RA it must be metabolized and eliminated. Three cytochromes (Cyp26A1, Cyp26B1 Cyp26C1) catalyze the oxidation of RA. The genes for these proteins are induced by high concentrations of RA, thus providing a regulatory feedback mechanism.[5]
Embryology
In vertebrates and invertebrate chordates, RA has a pivotal role during development. Altering levels of endogenous RA signaling during early embryology, both too low and too high, leads to birth defects,[25][26] including congenital vascular and cardiovascular defects.[27][28] Of note, fetal alcohol spectrum disorder encompasses congenital anomalies, including craniofacial, auditory, and ocular defects, neurobehavioral anomalies and mental disabilities caused by maternal consumption of alcohol during pregnancy. It is proposed that in the embryo there is competition between acetaldehyde, an ethanol metabolite, and retinaldehyde (retinal) for aldehyde dehydrogenase activity, resulting in a retenoic acid deficiency, and attributing the congenital birth defects to the loss of RA activated gene activation. In support of this theory, ethanol-induced developmental defects can be ameliorated by increasing the levels of retinol or retinal.[29] As for the risks of too much RA, the prescription drugs tretinoin (all-trans-retinoic acid) and isotretinoin (13-cis-retinoic acid), used orally or topically for acne treatment, come with warnings to not be used by pregnant women or women who are anticipating becoming pregnant, as they are known human teratogens.[30][31]
Immune functions
Vitamin A deficiency (VAD) has been linked to compromised resistance to infectious diseases.[32][33] In countries where early childhood VAD is common, vitamin A supplementation public health programs initiated in the 1980s were shown to reduce the incidence of diarrhea and measles, and all-cause mortality.[22][34][35] VAD also increases the risk of immune system over-reaction, leading to chronic inflammation in the intestinal system, stronger allergic reactions and autoimmune diseases.[32][33][36]
Lymphocytes and monocytes are types of white blood cells of the immune system.[37] Lymphocytes include natural killer cells, which function in innate immunity, T cells for adaptive cellular immunity and B cells for antibody-driven adaptive humoral immunity. Monocytes differentiate into macrophages and dendritic cells. Some lymphocytes migrate to the thymus where they differentiate into several types of T cells, in some instances referred to as "killer" or "helper" T cells and further differentiate after leaving the thymus. Each subtype has functions driven by the types of cytokines secreted and organs to which the cells preferentially migrate, also described as trafficking or homing.[38][39]
Reviews based on in vitro and animal research describe the role that retinoic acid (RA) has in the immune system. RA triggers receptors in bone marrow, resulting in generation of new white blood cells.[40] RA regulates proliferation and differentiation of white blood cells, the directed movement of T cells to the intestinal system, and to the up- and down-regulation of lymphocyte function.[32][33][34][35][36][41] If RA is adequate, T helper cell subtype Th1 is suppressed and subtypes Th2, Th17 and iTreg (for regulatory) are induced. Dendritic cells located in intestinal tissue have enzymes that convert retinal to all-trans retinoic acid, to be taken up by retinoic acid receptors on lymphocytes. The process triggers gene expression that leads to T cell types Th2, Th17 and iTreg moving to and taking up residence in mesenteric lymph nodes and Peyer's patches, respectively outside and on the inner wall of the small intestine.[34][35] The net effect is a down-regulation of immune activity, seen as tolerance of food allergens, and tolerance of resident bacteria and other organisms in the microbiome of the large intestine.[32][33][36] In a vitamin A deficient state, innate immunity is compromised and pro-inflammatory Th1 cells predominate.[32][41]
Skin
Deficiencies in vitamin A have been linked to an increased susceptibility to skin infection and inflammation.[42] Vitamin A appears to modulate the innate immune response and maintains homeostasis of epithelial tissues and mucosa through its metabolite, retinoic acid (RA). As part of the innate immune system, toll-like receptors in skin cells respond to pathogens and cell damage by inducing a pro-inflammatory immune response which includes increased RA production.[42] The epithelium of the skin encounters bacteria, fungi and viruses. Keratinocytes of the epidermal layer of the skin produce and secrete antimicrobial peptides (AMPs). Production of AMPs resistin and cathelicidin, are promoted by RA.[42]
Units of measurement
As some carotenoids can be converted into vitamin A, attempts have been made to determine how much of them in the diet is equivalent to a particular amount of retinol, so that comparisons can be made of the benefit of different foods. The situation can be confusing because the accepted equivalences have changed over time
For many years, a system of equivalencies in which an international unit (IU) was equal to 0.3 μg of retinol (~1 nmol), 0.6 μg of β-carotene, or 1.2 μg of other provitamin-A carotenoids was used.[43] This relationship was alternatively expressed by the retinol equivalent (RE): one RE corresponded to 1 μg retinol, to 2 μg β-carotene dissolved in oil, to 6 μg β-carotene in foods, and to 12 μg of either α-carotene, γ-carotene, or β-cryptoxanthin in food.
Newer research has shown that the absorption of provitamin-A carotenoids is only half as much as previously thought. As a result, in 2001 the US Institute of Medicine recommended a new unit, the retinol activity equivalent (RAE). Each μg RAE corresponds to 1 μg retinol, 2 μg of β-carotene in oil, 12 μg of "dietary" beta-carotene, or 24 μg of the three other dietary provitamin-A carotenoids.[4]
| Substance and its chemical environment (per 1 μg) | IU (1989) | μg RE (1989)[4] | μg RAE (2001)[4] |
|---|---|---|---|
| Retinol | 3.33 | 1 | 1 |
| beta-Carotene, dissolved in oil | 1.67 | 1/2 | 1/2 |
| beta-Carotene, common dietary | 1.67 | 1/6 | 1/12 |
|
0.83 | 1/12 | 1/24 |
Animal models have shown that at the enterocyte cell wall, β-carotene is taken up by the membrane transporter protein scavenger receptor class B, type 1 (SCARB1). Absorbed β-carotene is converted to retinal and then retinol. The first step of the conversion process consists of one molecule of β-carotene cleaved by the enzyme β-carotene-15, 15'-monooxygenase, which in humans and other mammalian species is encoded by the BCM01 gene,[7] into two molecules of retinal. When plasma retinol is in the normal range, gene expression for SCARB1 and BC01 are suppressed, creating a feedback loop that suppresses β-carotene absorption and conversion.[11] Absorption suppression is not complete, as receptor 36 is not downregulated.[11]
Dietary recommendations
The US National Academy of Medicine updated Dietary Reference Intakes (DRIs) in 2001 for vitamin A, which included Recommended Dietary Allowances (RDAs).[4] For infants up to 12 months there was not sufficient information to establish a RDA, so Adequate Intake (AI) is shown instead. As for safety, tolerable upper intake levels (ULs) were also established. For ULs, carotenoids are not added when calculating total vitamin A intake for safety assessments.[4]
| Life stage group | US RDAs or AIs (μg RAE/day)[4] |
US Upper limits (μg/day)[4] | |
|---|---|---|---|
| Infants | 0–6 months | 400 (AI) | 600 |
| 7–12 months | 500 (AI) | 600 | |
| Children | 1–3 years | 300 | 600 |
| 4–8 years | 400 | 900 | |
| Males | 9–13 years | 600 | 1700 |
| 14–18 years | 900 | 2800 | |
| >19 years | 900 | 3000 | |
| Females | 9–13 years | 600 | 1700 |
| 14–18 years | 700 | 2800 | |
| >19 years | 700 | 3000 | |
| Pregnancy | <19 years | 750 | 2800 |
| >19 years | 770 | 3000 | |
| Lactation | <19 years | 1200 | 2800 |
| >19 years | 1300 | 3000 | |
The European Food Safety Authority (EFSA) refers to the collective set of information as Dietary Reference Values, with Population Reference Intake (PRI) instead of RDA, and Average Requirement instead of EAR. AI and UL defined the same as in United States. For women and men of ages 15 and older, the PRIs are set respectively at 650 and 750 μg RE/day. PRI for pregnancy is 700 μg RE/day, for lactation 1300/day. For children of ages 1–14 years, the PRIs increase with age from 250 to 600 μg RE/day. These PRIs are similar to the US RDAs.[44] The EFSA reviewed the same safety question as the United States, and set ULs at 800 for ages 1–3, 1100 for ages 4–6, 1500 for ages 7–10, 2000 for ages 11–14, 2600 for ages 15–17 and 3000 μg/day for ages 18 and older for preformed vitamin A, i.e., not including dietary contributions from carotenoids.[45]
Safety
Vitamin A toxicity hypervitaminosis A occurs when too much vitamin A accumulates in the body. It comes from consumption of preformed vitamin A but not of carotenoids, as conversion of the latter to retinol is suppressed by the presence of adequate retinol.
Retinol safety
There are historical reports of acute hypervitaminosis from Arctic explorers consuming bearded seal or polar bear liver, both very rich sources of stored retinol,[46] and there are also case reports of acute hypervitaminosis from consuming fish liver,[47] but otherwise there is no risk from consuming too much via commonly consumed foods. Only consumption of retinol-containing dietary supplements can result in acute or chronic toxicity.[5] Acute toxicity occurs after a single or short-term doses of greater than 150,000 μg. Symptoms include blurred vision, nausea, vomiting, dizziness and headache within 8 to 24 hours. For infants ages 0–6 months given an oral dose to prevent development of vitamin A deficiency, bulging skull fontanel was evident after 24 hours, usually resolved by 72 hours.[48] Chronic toxicity may occur with long-term consumption of vitamin A at doses of 25,000–33,000 IU/day for several months.[3] Excessive consumption of alcohol can lead to chronic toxicity at lower intakes.[2] Symptoms may include nervous system effects, liver abnormalities, fatigue, muscle weakness, bone and skin changes and others. The adverse effects of both acute and chronic toxicity are reversed after consumption is stopped.[4]
In 2001, for the purpose of determining ULs for adults, the US Institute of Medicine considered three primary adverse effects and settled on two: teratogenicity, i.e., causing birth defects, and liver abnormalities. Reduced bone mineral density was considered, but dismissed because the human evidence was contradictory.[4] During pregnancy, especially during the first trimester, consumption of retinol in amounts exceeding 4,500 μg/day increased the risk of birth defects, but not below that amount, thus setting a "No-Observed Adverse-Effect Level" (NOAEL). Given the quality of the clinical trial evidence, the NOAEL was divided by an uncertainty factor of 1.5 to set the UL for women of reproductive age at 3,000 μg/day of preformed vitamin A. For all other adults, liver abnormalities were detected at intakes above 14,000 μg/day. Given the weak quality of the clinical evidence, an uncertainty factor of 5 was used, and with rounding, the UL was set at 3,000 μg/day. Despite a US UL set at 3,000 μg, it is possible to buy over-the-counter dietary supplement products which are 7,500 μg (25,000 IU), with a label caution statement "Not intended for long term use unless under medical supervision."[49]
For children ULs were extrapolated from the adult value, adjusted for relative body weight. For infants, several case studies reported adverse effects that include bulging fontanels, increased intracranial pressure, loss of appetite, hyperirritability and skin peeling after chronic ingestion of the order of 6,000 or more μg/day. Given the small database, an uncertainty factor of 10 divided into the "Lowest-Observed-Adverse-Effect Level" (LOAEL) led to a UL of 600 μg/day.[4]
β-carotene safety
No adverse effects other than carotenemia have been reported for consumption of β-carotene rich foods. Supplementation with β-carotene does not cause hypervitaminosis A.[11] Two large clinical trials (ATBC and CARET) were conducted in tobacco smokers to see if years of β-carotene supplementation at 20 or 30 mg/day in oil-filled capsules would reduce the risk of lung cancer.[50] These trials were implemented because observational studies had reported a lower incidence of lung cancer in tobacco smokers who had diets higher in β-carotene. Unexpectedly, this high-dose β-carotene supplementation resulted in a higher incidence of lung cancer and of total mortality.[11] Taking this and other evidence into consideration, the U.S. Institute of Medicine decided to not set a Tolerable Upper Intake Level (UL) for β-carotene.[11][50] The European Food Safety Authority, acting for the European Union, also decided to not set a UL for β-carotene.[45]

Carotenosis
Carotenoderma, also referred to as carotenemia, is a benign and reversible medical condition where an excess of dietary carotenoids results in orange discoloration of the outermost skin layer. It is associated with a high blood β-carotene value. This can occur after a month or two of consumption of beta-carotene rich foods, such as carrots, carrot juice, tangerine juice, mangos, or in Africa, red palm oil. β-carotene dietary supplements can have the same effect. The discoloration extends to palms and soles of feet, but not to the white of the eye, which helps distinguish the condition from jaundice.[51] Consumption of greater than 30 mg/day for a prolonged period has been confirmed as leading to carotenemia.[11][52]
U.S. labeling
For U.S. food and dietary supplement labeling purposes, the amount in a serving is expressed as a percent of Daily Value (%DV). For vitamin A labeling purposes 100% of the Daily Value was set at 5,000 IU, but it was revised to 900 μg RAE on 27 May 2016.[53][54] A table of the old and new adult daily values is provided at Reference Daily Intake.
Sources
| Food | Retinol
Micrograms per 100 g[55] |
|---|---|
| cod liver oil | 30000 |
| beef liver (cooked) | 21145 |
| chicken liver (cooked) | 4296 |
| butter (stick) | 684 |
| cheddar cheese | 316 |
| egg (cooked) | 140 |
Vitamin A is found in many foods.[55] Vitamin A in food exists either as preformed retinol – an active form of vitamin A – found in animal liver, dairy and egg products, and some fortified foods, or as provitamin A carotenoids, which are plant pigments digested into vitamin A after consuming carotenoid-rich plant foods, typically in red, orange, or yellow colors.[3] Carotenoid pigments may be masked by chlorophylls in dark green leaf vegetables, such as spinach. The relatively low bioavailability of plant-food carotenoids results partly from binding to proteins – chopping, homogenizing or cooking disrupts the plant proteins, increasing provitamin A carotenoid bioavailability.[3]
Vegetarian and vegan diets can provide sufficient vitamin A in the form of provitamin A carotenoids if the diet contains green leafy vegetables such as spinach and kale, carrots, carrot juice, sweet potatoes and other carotenoid-rich foods. In the U.S., the average daily intake of β-carotene is in the range 2–7 mg.[56]
Some manufactured foods and dietary supplements are sources of vitamin A or beta-carotene.[3][4]
Despite the US setting an adult upper limit of 3,000 μg/day, some companies sell vitamin A as a dietary supplement with amounts of 7,500 μg/day. Two examples are WonderLabs and Pure Prescriptions.[57][58]
Fortification
Some countries require or recommend fortification of foods. As of January 2022, 37 countries, mostly in Sub-Saharan Africa, require food fortification of cooking oil, rice, wheat flour or maize (corn) flour with vitamin A, usually as retinyl palmitate or retinyl acetate. Examples include Pakistan, oil, 11.7 mg/kg and Nigeria, oil, 6 mg/kg; wheat and maize flour, 2 mg/kg.[59] An additional 12 countries, mostly in southeast Asia have a voluntary fortification program. For example, the government of India recommends 7.95 mg/kg in oil and 0.626 mg/kg for wheat flour and rice. However, compliance in countries with voluntary fortification is lower than countries with mandatory fortification.[59] No countries in Europe or North America fortify foods with vitamin A.[59]
| Food | Beta-carotene
Milligrams per 100 g[55] |
|---|---|
| Sweet potato, skinned, boiled | 9.4 |
| Carrot juice | 9.3 |
| Carrots, raw or boiled | 9.2 |
| Kale, boiled | 8.8 |
| Pumpkin, canned | 6.9 |
| Spinach, canned | 5.9 |
Separated from fortification via addition of synthetic vitamin A to foods, means of fortifying foods via genetic engineering have been explored. Research on rice began in 1982.[60] The first field trials of golden rice cultivars were conducted in 2004.[61] The result was "Golden Rice", a variety of Oryza sativa rice produced through genetic engineering to biosynthesize beta-carotene, a precursor of retinol, in the edible parts of rice.[62][63] In May 2018, regulatory agencies in the United States, Canada, Australia and New Zealand had concluded that Golden Rice met food safety standards.[64] On 21 July 2021, the Philippines became the first country to officially issue the biosafety permit for commercially propagating Golden Rice.[65][66]
%252C_OWID.svg.png.webp)
Vitamin A supplementation (VAS)
Delivery of oral high-dose supplements remains the principal strategy for minimizing deficiency.[68]
As of 2017, more than 80 countries worldwide are implementing universal VAS programs targeted to children 6–59 months of age through semi-annual national campaigns.[69]
Doses in these programs are one dose of 50,000 or 100,000 IU for children aged 6 to 11 months and 100,000 to 200,000 IU for children aged 12 months to five years, every four to six months.[22]
Deficiency
Primary causes

Vitamin A deficiency is common in developing countries, especially in Sub-Saharan Africa and Southeast Asia. Deficiency can occur at any age, but is most common in pre-school-age children and pregnant women, the latter due to a need to transfer retinol to the fetus. The causes are low intake of retinol-containing, animal-sourced foods and low intake of carotene-containing, plant-sourced foods. Vitamin A deficiency is estimated to affect approximately one third of children under the age of five around the world,[70] possibly leading to the deaths of 670,000 children under five annually.[71]
Between 250,000 and 500,000 children in developing countries become blind each year owing to vitamin A deficiency.[2] Vitamin A deficiency is "the leading cause of preventable childhood blindness", according to UNICEF.[9][72] It also increases the risk of death from common childhood conditions, such as diarrhea. UNICEF regards addressing vitamin A deficiency as critical to reducing child mortality, the fourth of the United Nations' Millennium Development Goals.[9]
During diagnosis, night blindness and dry eyes are signs of vitamin A deficiency that can be recognized without requiring biochemical tests. Plasma retinol is used to confirm vitamin A status. A plasma concentration of about 2.0 μmol/L is normal; less than 0.70 μmol/L (equivalent to 20 μg/dL) indicates moderate vitamin A deficiency, and less than 0.35 μmol/L (10 μg/dL) indicates severe vitamin A deficiency. Breast milk retinol of less than 8 μg/gram milk fat is considered insufficient.[5] One weakness of these measures is that they are not good indicators of liver vitamin A stores as retinyl esters in hepatic stellate cells. The amount of vitamin A leaving the liver, bound to retinol binding protein (RBP), is under tight control as long as there are sufficient liver reserves. Only when liver content of vitamin A drops below approximately 20 μg/gram will concentration in the blood decline.[4][73]
Secondary causes
There are causes for deficiency other than low dietary intake of vitamin A as retinol or carotenes. Inadequate dietary protein and caloric energy are needed for a normal rate of synthesis of RBP, without which, retinol cannot be mobilized to leave the liver. Systemic infections can cause transient decreases in RBP synthesis even if protein-calorie malnutrition is absent. Chronic alcohol consumption reduces liver vitamin A storage.[4] Non-alcoholic fatty liver disease (NAFLD), characterized by the accumulation of fat in the liver, is the hepatic manifestation of metabolic syndrome. Liver damage from NAFLD reduces liver storage capacity for retinol and reduces the ability to mobilize liver stores to maintain normal circulating concentration.[74]
Animal requirements
All vertebrate and chordate species require vitamin A,[26] either as dietary carotenoids or preformed retinol from consuming other animals. Deficiencies have been reported in laboratory-raised and pet dogs, cats, birds, reptiles and amphibians,[75][76] also commercially raised chickens and turkeys.[77] Herbivore species such as horses, cattle and sheep can get sufficient β-carotene from green pasture to be healthy, but the content in pasture grass dry due to drought and long-stored hay can be too low, leading to vitamin A deficiency.[75] Omnivore and carnivore species, especially those toward the top of the food chain, can accrue large amounts of retinyl esters in their livers, or else excrete retinyl esters in urine as a means of dealing with surplus.[15] Before the era of synthetic retinol, cod liver oil, high in vitamins A and D, was a commonly consumed dietary supplement.[78][79] Invertebrates cannot synthesize carotenoids or retinol, and thus must accrue these essential nutrients from consumption of algae, plants or animals.[80][81][82]
Medical uses
Preventing and treating deficiency
Recognition of its prevalence and consequences has led to governments and non-government organizations promoting vitamin A fortification of foods[59] and creating programs that administer large bolus-size oral doses of vitamin A to young children every four to six months.[69] In 2008, the World Health Organization estimated that vitamin A supplementation over a decade in 40 countries averted 1.25 million deaths due to vitamin A deficiency.[83] A Cochrane review reported that vitamin A supplementation is associated with a clinically meaningful reduction in morbidity and mortality in children ages six month to five years of age. All-cause mortality was reduced by 14%, and incidences of diarrhea by 12%.[22] However, a Cochrane review by the same group concluded there was insufficient evidence to recommend blanket vitamin A supplementation for infants one to six months of age, as it did not reduce infant mortality or morbidity.[48]
Oral retinoic acid
Orally consumed retinoic acid (RA), as all-trans-tretinoin or 13-cis-isotretinoin has been shown to improve facial skin health by switching on genes and differentiating keratinocytes (immature skin cells) into mature epidermal cells. RA reduces the size and secretion of the sebaceous glands, and by doing so reduces bacterial numbers in both the ducts and skin surface. It reduces inflammation via inhibition of chemotactic responses of monocytes and neutrophils. In the US, isotretinoin was released to the market in 1982 as a revolutionary treatment for severe and refractory acne vulgaris. It was shown that a dose of 0.5‑1.0 mg/kg body weight/day is enough to produce a reduction in sebum excretion by 90% within a month or two, but the recommended treatment duration is 4 to 6 months.[30] Isotretinoin is a known teratogen, with an estimated 20‑35% risk of physical birth defects to infants that are exposed to isotretinoin in utero, including numerous congenital defects such as craniofacial defects, cardiovascular and neurological malformations or thymic disorders. Neurocognitive impairments in the absence of any physical defects has been established to be 30‑60%. For these reasons, physician- and patient-education programs were initiated, recommending that for women of child-bearing age, contraception be initiated a month before starting oral (or topical) isotretinoin, and continue for a month after treatment ended.[30]
In addition to the approved use for treating acne vulgaris, researchers have investigated off-label applications for dermatological conditions, such as rosacea, psoriasis, and other conditions.[84] Rosacea was reported as responding favorably to doses lower than used for acne. Isotretinoin in combination with ultraviolet light was shown affective for treating psoriasis. Isotretinoin in combination with injected interferon-alpha showed some potential for treating genital warts. Isotretinoin in combination with topical fluorouracil or injected interferon-alpha showed some potential for treating precancerous skin lesions and skin cancer.[84]
Topical retinoic acid and retinol
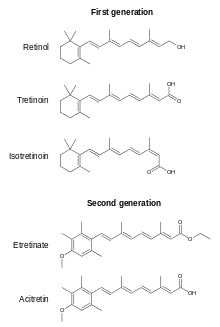
Retinoic acids tretinoin (all-trans-retinoic acid) and isotretinoin (13-cis-retinoic acid) are prescription topical medications used to treat moderate to severe cystic acne and acne not responsive to other treatments.[85][86][87][88] These are usually applied as a skin cream to the face after cleansing to remove make-up and skin oils. Tretinoin and isotretinoin act by binding to two nuclear receptor families within keratinocytes: the retinoic acid receptors (RAR) and the retinoid X receptors (RXR).[89] These events contribute to the normalization of follicular keratinization and decreased cohesiveness of keratinocytes, resulting in reduced follicular occlusion and microcomedone formation.[90] The retinoid-receptor complex competes for coactivator proteins of AP-1, a key transcription factor involved in inflammation.[89] Retinoic acid products also reduce sebum secretion, a nutrient source for bacteria, from facial pores.
These drugs are US-designated Pregnancy Category C (animal reproduction studies have shown an adverse effect on the fetus), and should not be used by pregnant women or women who are anticipating becoming pregnant.[31] Many countries established a physician- and patient- education pregnancy prevention policy.[91]
Trifarotene is a prescription retinoid for the topical treatment acne vulgaris.[13] It functions as a retinoic acid receptor (RAR)-γ agonist.[92]
Non-prescription topical products that have health claims for reducing facial acne, combating skin dark spots and reducing wrinkles and lines associated with aging often contain retinyl palmitate. The hypothesis is that this is absorbed and desterified to free retinol, then converted to retinaldehyde and further metabolized to all-trans retinoic acid, whence it will have the same effects as prescription products with fewer side effects.[93] There is some ex vivo evidence with human skin that esterified retinol is absorbed and then converted to retinol.[94] In addition to esterified retinol, some of these products contain hydroxypinacolone retinoate, identified as esterified 9-cis-retinoic acid.[95]
Synthesis
Biosynthesis
Carotenoid synthesis takes place in plants, certain fungi, and bacteria. Structurally carotenes are tetraterpenes, meaning that they are synthesized biochemically from four 10-carbon terpene units, which in turn were formed from eight 5-carbon isoprene units. Intermediate steps are the creation of a 40-carbon phytoene molecule, conversion to lycopene via desaturation, and then creation of ionone rings at both ends of the molecule. β-carotene has a β-ionone ring at both ends, meaning that the molecule can be divided symmetrically to yield two retinol molecules. α-Carotene has a β-ionone ring at one end and an Ɛ-ionone ring at the other, so it has half the retinol conversion capacity.[11]
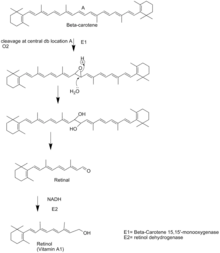
In most animal species, retinol is synthesized from the breakdown of the plant-formed provitamin, β-carotene. First, the enzyme beta-carotene 15,15'-dioxygenase (BCO-1) cleaves β-carotene at the central double bond, creating an epoxide. This epoxide is then attacked by water creating two hydroxyl groups in the center of the structure. The cleavage occurs when these alcohols are reduced to the aldehydes using NADH. The resultant retinal is then quickly reduced to retinol by the enzyme retinol dehydrogenase.[5] Omnivore species such as dogs, wolves, coyotes and foxes in general are low producers of BCO-1. The enzyme is lacking in felids (cats), meaning that vitamin A requirements are met from the retinyl ester content of prey animals.[15]
Industrial synthesis
β-carotene can be extracted from fungus Blakeslea trispora, marine algae Dunaliella salina or genetically modified yeast Saccharomyces cerevisiae, starting with xylose as a substrate.[96] Chemical synthesis uses either a method developed by BASF[97][98] or a Grignard reaction utilized by Hoffman-La Roche.[99]
The world market for synthetic retinol is primarily for animal feed, leaving approximately 13% for a combination of food, prescription medication and dietary supplement use.[100] Industrial methods for the production of retinol rely on chemical synthesis. The first industrialized synthesis of retinol was achieved by the company Hoffmann-La Roche in 1947. In the following decades, eight other companies developed their own processes. β-ionone, synthesized from acetone, is the essential starting point for all industrial syntheses. Each process involves elongating the unsaturated carbon chain.[100] Pure retinol is extremely sensitive to oxidization and is prepared and transported at low temperatures and oxygen-free atmospheres. When prepared as a dietary supplement or food additive, retinol is stabilized as the ester derivatives retinyl acetate or retinyl palmitate. Prior to 1999, three companies, Roche, BASF and Rhone-Poulenc controlled 96% of global vitamin A sales. In 2001, the European Commission imposed total fines of 855.22 million euros on these and five other companies for their participation in eight distinct market-sharing and price-fixing cartels that dated back to 1989.[101] Roche sold its vitamin division to DSM in 2003. DSM and BASF have the major share of industrial production.[100] A biosynthesis alternative utilizes genetically engineered yeast species Saccharomyces cerevisiae to synthesize retinal and retinol, using xylose as a starting substrate. This was accompished by having the yeast first synthesize β-carotene and then the cleaving enzyme β-carotene 15,15'-dioxygenase to yield retinal.[102]
Research
Cancer
Meta-analyses of intervention and observational trials for various types of cancer report mixed results. Supplementation with β-carotene did not appear to decrease the risk of cancer overall, nor specific cancers including: pancreatic, colorectal, prostate, breast, melanoma, or skin cancer generally.[103] High-dose β-carotene supplementation unexpectedly resulted in a higher incidence of lung cancer and of total mortality in people who were cigarette smokers.[11]
For dietary retinol, no effects were observed for high dietary intake and breast cancer survival,[104] risk of liver cancer,[105] risk of bladder cancer[106] or risk of colorectal cancer,[107][108] although the last review did report lower risk for higher beta-carotene consumption.[108] In contrast, an inverse association was reported between retinol intake and relative risk of esophageal cancer,[109] gastric cancer,[110] ovarian cancer,[111] pancreatic cancer,[112] lung cancer,[113] melanoma,[114] and cervical cancer.[115] For lung cancer, an inverse association was also seen for beta-carotene intake, separate from the retinol results.[113] When high dietary intake was compared to low dietary intake, the decreases in relative risk were in the range of 15 to 20%. For gastric cancer, a meta-analysis of prevention trials reported a 29% decrease in relative risk from retinol supplementation at 1500 μg/day.[116]
Fetal alcohol spectrum disorder
Fetal alcohol spectrum disorder (FASD), formerly referred to as fetal alcohol syndrome, presents as craniofacial malformations, neurobehavioral disorders and mental disabilities, all attributed to exposing human embryos to alcohol during fetal development.[117][118] The risk of FASD depends on the amount consumed, the frequency of consumption, and the points in pregnancy at which the alcohol is consumed.[119] Ethanol is a known teratogen, i.e., causes birth defects. Ethanol is metabolized by alcohol dehydrogenase enzymes into acetaldehyde.[120][121] The subsequent oxidation of acetaldehyde into acetate is performed by aldehyde dehydrogenase enzymes. Given that retinoic acid (RA) regulates numerous embryonic and differentiation processes, one of the proposed mechanisms for the teratogenic effects of ethanol is a competition for the enzymes required for the biosynthesis of RA from vitamin A. Animal research demonstrates that in the embryo, the competition takes place between acetaldehyde and retinaldehyde for aldehyde dehydrogenase activity. In this model, acetaldehyde inhibits the production of retinoic acid by retinaldehyde dehydrogenase. Ethanol-induced developmental defects can be ameliorated by increasing the levels of retinol, retinaldehyde, or retinaldehyde dehydrogenase. Thus, animal research supports the reduction of retinoic acid activity as an etiological trigger in the induction of FASD.[117][118][122][123]
Malaria
Malaria and vitamin A deficiency are both common among young children in sub-Saharan Africa. Vitamin A supplementation to children in regions where vitamin A deficiency is common has repeatedly been shown to reduce overall mortality rates, especially from measles and diarrhea.[124] For malaria, clinical trial results are mixed, either showing that vitamin A treatment did not reduce the incidence of probable malarial fever, or else did not affect incidence, but did reduce slide-confirmed parasite density and reduced the number of fever episodes.[124] The question was raised as to whether malaria causes vitamin A deficiency, or vitamin A deficiency contributes to the severity of malaria, or both. Researchers proposed several mechanisms by which malaria (and other infections) could contribute to vitamin A deficiency, including a fever-induced reduction in synthesis of retinal-binding protein (RBP) responsible for transporting retinol from liver to plasma and tissues, but reported finding no evidence for a transient depression or restoration of plasma RBP or retinol after a malarial infection was eliminated.[124]
History

In 1912, Frederick Gowland Hopkins demonstrated that unknown accessory factors found in milk, other than carbohydrates, proteins, and fats were necessary for growth in rats. Hopkins received a Nobel Prize for this discovery in 1929.[6][125] By 1913, one of these substances was independently discovered by Elmer McCollum and Marguerite Davis at the University of Wisconsin–Madison, and Lafayette Mendel and Thomas Burr Osborne at Yale University. McCollum and Davis ultimately received credit because they submitted their paper three weeks before Mendel and Osborne. Both papers appeared in the same issue of the Journal of Biological Chemistry in 1913.[126] The "accessory factors" were termed "fat soluble" in 1918, and later "vitamin A" in 1920. In 1919, Harry Steenbock (University of Wisconsin–Madison) proposed a relationship between yellow plant pigments (beta-carotene) and vitamin A. In 1931, Swiss chemist Paul Karrer described the chemical structure of vitamin A.[125] Retinoic acid and retinol were first synthesized in 1946 and 1947 by two Dutch chemists, David Adriaan van Dorp and Jozef Ferdinand Arens.[127][128]

During World War II, German bombers would attack at night to evade British defenses. In order to keep the 1939 invention of a new on-board Airborne Intercept Radar system secret from German bombers, the British Ministry of Information told newspapers that the nighttime defensive success of Royal Air Force pilots was due to a high dietary intake of carrots rich in beta-carotene, propagating the myth that carrots enable people to see better in the dark.[129]
In 1967, George Wald shared the Nobel Prize in Physiology and Medicine for his work on chemical visual processes in the eye.[130] Wald had demonstrated in 1935 that photoreceptor cells in the eye contain rhodopsin, a chromophore composed of the protein opsin and 11-cis retinal. When struck by light, 11-cis retinal undergoes photoisomerization to all-trans retinal and via signal transduction cascade send a nerve signal to the brain. The all-trans retinal is reduced to all-trans retinol and travels back to the retinal pigment epithelium to be recycled to 11-cis retinal and reconjugated to opsin.[6][131] Wald's work was the culmination of nearly 60 years of research. In 1877, Franz Christian Boll identified a light-sensitive pigment in the outer segments of rod cells of the retina that faded/bleached when exposed to light, but was restored after light exposure ceased. He suggested that this substance, by a photochemical process, conveyed the impression of light to the brain.[6] The research was taken up by Wilhelm Kühne, who named the pigment rhodopsin, also known as "visual purple." Kühne confirmed that that rhodopsin is extremely sensitive to light, and thus enables vision in low-light conditions, and that it was this chemical decomposition that stimulated nerve impulses to the brain.[6] Research stalled until after identification of "fat-soluble vitamin A" as a dietary substance found in milkfat but not lard, would reverse night blindness and xerophthalmia. In 1925, Fridericia and Holm demonstrated that vitamin A deficient rats were unable to regenerate rhodopsin after being moved from a light to a dark room.[132]
References
- 1 2 "Vitamin A". The American Society of Health-System Pharmacists. Archived from the original on 30 December 2016. Retrieved 8 December 2016.
- 1 2 3 "Vitamin A Fact Sheet for Health Professionals". Office of Dietary Supplements, US National Institutes of Health. March 2021. Archived from the original on 12 December 2016. Retrieved 8 August 2021.
- 1 2 3 4 5 6 7 8 "Vitamin A". Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis. 1 July 2016. Archived from the original on 27 April 2021. Retrieved 21 December 2021.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Institute of Medicine (2001). "Vitamin A". Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Food and Nutrition Board of the Institute of Medicine. pp. 82–161. ISBN 0-309-07290-5. Archived from the original on 28 April 2021. Retrieved 8 June 2022.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Blaner WS (2020). "Vitamin A". In BP Marriott, DF Birt, VA Stallings, AA Yates (eds.). Present Knowledge in Nutrition, Eleventh Edition. London, United Kingdom: Academic Press (Elsevier). pp. 73–92. ISBN 978-0-323-66162-1.
- 1 2 3 4 5 6 Wolf G (June 2001). "The discovery of the visual function of vitamin A". The Journal of Nutrition. 131 (6): 1647–50. doi:10.1093/jn/131.6.1647. PMID 11385047.
- 1 2 3 Wu L, Guo X, Wang W, Medeiros DM, Clarke SL, Lucas EA, Smith BJ, Lin D (November 2016). "Molecular aspects of β, β-carotene-9', 10'-oxygenase 2 in carotenoid metabolism and diseases". Exp Biol Med (Maywood). 241 (17): 1879–87. doi:10.1177/1535370216657900. PMC 5068469. PMID 27390265.
- 1 2 Kedishvili NY (2016). "Retinoic Acid Synthesis and Degradation". Subcell Biochem. Subcellular Biochemistry. 81: 127–61. doi:10.1007/978-94-024-0945-1_5. ISBN 978-94-024-0943-7. PMC 5551983. PMID 27830503.
- 1 2 3 "Vitamin A Deficiency" Archived 12 September 2016 at the Wayback Machine, UNICEF. Retrieved 3 June 2015.
- ↑ Lidén M, Eriksson U (May 2006). "Understanding retinol metabolism: structure and function of retinol dehydrogenases". The Journal of Biological Chemistry. 281 (19): 13001–13004. doi:10.1074/jbc.R500027200. PMID 16428379.
- 1 2 3 4 5 6 7 8 9 von Lintig J (2020). "Carotenoids". In BP Marriott, DF Birt, VA Stallings, AA Yates (eds.). Present Knowledge in Nutrition, Eleventh Edition. London, United Kingdom: Academic Press (Elsevier). pp. 531–49. ISBN 978-0-323-66162-1.
- 1 2 Beckenbach L, Baron JM, Merk HF, Löffler H, Amann PM (2015). "Retinoid treatment of skin diseases". Eur J Dermatol. 25 (5): 384–91. doi:10.1684/ejd.2015.2544. PMID 26069148. S2CID 408725.
- 1 2 "Trifarotene Monograph for Professionals". Drugs.com. 28 October 2019. Archived from the original on 4 March 2021. Retrieved 23 May 2021.
- 1 2 Chelstowska S, Widjaja-Adhi MA, Silvaroli JA, Golczak M (October 2016). "Molecular Basis for Vitamin A Uptake and Storage in Vertebrates". Nutrients. 8 (11): 676. doi:10.3390/nu8110676. PMC 5133064. PMID 27792183.
- 1 2 3 4 5 6 Green AS, Fascetti AJ (2016). "Meeting the Vitamin A Requirement: The Efficacy and Importance of β-Carotene in Animal Species". ScientificWorldJournal. 2016: 7393620. doi:10.1155/2016/7393620. PMC 5090096. PMID 27833936.
- ↑ Roberts C (March 2020). "Regulating Retinoic Acid Availability during Development and Regeneration: The Role of the CYP26 Enzymes". J Dev Biol. 8 (1): 6. doi:10.3390/jdb8010006. PMC 7151129. PMID 32151018.
- ↑ Isoherranen N, Zhong G (December 2019). "Biochemical and physiological importance of the CYP26 retinoic acid hydroxylases". Pharmacol Ther. 204: 107400. doi:10.1016/j.pharmthera.2019.107400. PMC 6881548. PMID 31419517.
- 1 2 3 4 Whitcher JP, Srinivasan M, Upadhyay MP (2001). "Corneal blindness: a global perspective". Bull World Health Organ. 79 (3): 214–21. PMC 2566379. PMID 11285665.
- 1 2 Combs GF (2008). The Vitamins: Fundamental Aspects in Nutrition and Health (3rd ed.). Burlington, MA: Elsevier Academic Press. ISBN 978-0-12-183493-7.
- 1 2 Miller RE, Tredici TJ (1 August 1992). "Night Vision Manual for the Flight Surgeon". US Department of Defense, Defense Technical Information Center. Archived from the original on 4 January 2022. Retrieved 4 January 2022.
- 1 2 3 Akhtar S, Ahmed A, Randhawa MA, Atukorala S, Arlappa N, Ismail T, Ali Z (December 2013). "Prevalence of vitamin A deficiency in South Asia: causes, outcomes, and possible remedies". J Health Popul Nutr. 31 (4): 413–23. doi:10.3329/jhpn.v31i4.19975. PMC 3905635. PMID 24592582.
- 1 2 3 4 Imdad A, Mayo-Wilson E, Herzer K, Bhutta ZA (March 2022). "Vitamin A supplementation for preventing morbidity and mortality in children from six months to five years of age". Cochrane Database Syst Rev. 3 (11): CD008524. doi:10.1002/14651858.CD008524.pub4. PMC 8925277. PMID 35294044.
{{cite journal}}: CS1 maint: PMC embargo expired (link) - ↑ Duester G (September 2008). "Retinoic acid synthesis and signaling during early organogenesis". Cell. 134 (6): 921–31. doi:10.1016/j.cell.2008.09.002. PMC 2632951. PMID 18805086.
- ↑ Stipanuk MH (2006). Biochemical, Physiological and Molecular Aspects of Human Nutrition (2nd ed.). Philadelphia: Saunders. ISBN 9781416002093.
- ↑ Metzler MA, Sandell LL (December 2016). "Enzymatic Metabolism of Vitamin A in Developing Vertebrate Embryos". Nutrients. 8 (12): 812. doi:10.3390/nu8120812. PMC 5188467. PMID 27983671.
- 1 2 Marlétaz F, Holland LZ, Laudet V, Schubert M (2006). "Retinoic acid signaling and the evolution of chordates". Int J Biol Sci. 2 (2): 38–47. doi:10.7150/ijbs.2.38. PMC 1458431. PMID 16733532.
- ↑ Pawlikowski B, Wragge J, Siegenthaler JA (July 2019). "Retinoic acid signaling in vascular development". Genesis. 57 (7–8): e23287. doi:10.1002/dvg.23287. PMC 6684837. PMID 30801891.
- ↑ Wang S, Moise AR (July 2019). "Recent insights on the role and regulation of retinoic acid signaling during epicardial development". Genesis. 57 (7–8): e23303. doi:10.1002/dvg.23303. PMC 6682438. PMID 31066193.
- ↑ Shabtai Y, Fainsod A (April 2018). "Competition between ethanol clearance and retinoic acid biosynthesis in the induction of fetal alcohol syndrome". Biochem Cell Biol. 96 (2): 148–160. doi:10.1139/bcb-2017-0132. PMID 28982012.
- 1 2 3 Draghici CC, Miulescu RG, Petca RC, Petca A, Dumitrașcu MC, Șandru F (May 2021). "Teratogenic effect of isotretinoin in both fertile females and males (Review)". Exp Ther Med. 21 (5): 534. doi:10.3892/etm.2021.9966. PMC 8014951. PMID 33815607.
- 1 2 "Tretinoin topical Use During Pregnancy". Drugs.com. 1 July 2019. Archived from the original on 29 November 2020. Retrieved 16 January 2020.
- 1 2 3 4 5 Ross AC (November 2012). "Vitamin A and retinoic acid in T cell-related immunity". Am J Clin Nutr. 96 (5): 1166S–72S. doi:10.3945/ajcn.112.034637. PMC 3471201. PMID 23053562.
- 1 2 3 4 Pino-Lagos K, Guo Y, Noelle RJ (2010). "Retinoic acid: a key player in immunity". Biofactors. 36 (6): 430–36. doi:10.1002/biof.117. PMC 3826167. PMID 20803520.
- 1 2 3 Brown CC, Noelle RJ (May 2015). "Seeing through the dark: New insights into the immune regulatory functions of vitamin A". Eur J Immunol. 45 (5): 1287–95. doi:10.1002/eji.201344398. PMC 4426035. PMID 25808452.
- 1 2 3 Guo Y, Brown C, Ortiz C, Noelle RJ (January 2015). "Leukocyte homing, fate, and function are controlled by retinoic acid". Physiol Rev. 95 (1): 125–48. doi:10.1152/physrev.00032.2013. PMC 4281589. PMID 25540140.
- 1 2 3 Bono MR, Tejon G, Flores-Santibañez F, Fernandez D, Rosemblatt M, Sauma D (June 2016). "Retinoic Acid as a Modulator of T Cell Immunity". Nutrients. 8 (6): 349. doi:10.3390/nu8060349. PMC 4924190. PMID 27304965.
- ↑ Janeway C, Travers P, Walport M, Shlomchik M (2001). Immunobiology (5th ed.). New York and London: Garland Science. ISBN 0-8153-4101-6. Archived from the original on 28 June 2009. Retrieved 8 June 2022.
- ↑ Omman RA, Kini AR (2020). "Leukocyte development, kinetics, and functions". In Keohane EM, Otto CN, Walenga JN (eds.). Rodak's Hematology: Clinical Principles and Applications (6th ed.). St. Louis, Missouri: Elsevier. pp. 117–35. ISBN 978-0-323-53045-3.
- ↑ Cohn L, Hawrylowicz C, Ray A (2014). "Biology of Lymphocytes". Middleton's Allergy: Principles and Practice (8th ed.). Philadelphia: Saunders. pp. 203–14. doi:10.1016/B978-0-323-08593-9.00013-9. ISBN 9780323085939. Archived from the original on 15 January 2022. Retrieved 8 June 2022.
- ↑ Cañete A, Cano E, Muñoz-Chápuli R, Carmona R (February 2017). "Role of Vitamin A/Retinoic Acid in Regulation of Embryonic and Adult Hematopoiesis". Nutrients. 9 (2): 159. doi:10.3390/nu9020159. PMC 5331590. PMID 28230720.
- 1 2 Czarnewski P, Das S, Parigi SM, Villablanca EJ (January 2017). "Retinoic Acid and Its Role in Modulating Intestinal Innate Immunity". Nutrients. 9 (1): 68. doi:10.3390/nu9010068. PMC 5295112. PMID 28098786.
- 1 2 3 Roche FC, Harris-Tryon TA (January 2021). "Illuminating the Role of Vitamin A in Skin Innate Immunity and the Skin Microbiome: A Narrative Review". Nutrients. 13 (2): 302. doi:10.3390/nu13020302. PMC 7909803. PMID 33494277.
- ↑ Composition of Foods Raw, Processed, Prepared USDA National Nutrient Database for Standard Reference, Release 20 USDA, Feb. 2008
- ↑ "Overview on Dietary Reference Values for the EU population as derived by the EFSA Panel on Dietetic Products, Nutrition and Allergies" (PDF). 2017. Archived (PDF) from the original on 28 August 2017. Retrieved 8 June 2022.
- 1 2 Tolerable Upper Intake Levels For Vitamins And Minerals (PDF), European Food Safety Authority, 2006, archived (PDF) from the original on 16 March 2016, retrieved 8 June 2022
- ↑ Rodahl K, Moore T (July 1943). "The vitamin A content and toxicity of bear and seal liver". Biochem J. 37 (2): 166–68. doi:10.1042/bj0370166. PMC 1257872. PMID 16747610.
- ↑ Schmitt C, Domangé B, Torrents R, de Haro L, Simon N (December 2020). "Hypervitaminosis A Following the Ingestion of Fish Liver: Report on 3 Cases from the Poison Control Center in Marseille". Wilderness Environ Med. 31 (4): 454–456. doi:10.1016/j.wem.2020.06.003. PMID 32861618. S2CID 221384282.
- 1 2 Imdad A, Ahmed Z, Bhutta ZA (September 2016). "Vitamin A supplementation for the prevention of morbidity and mortality in infants one to six months of age". The Cochrane Database of Systematic Reviews. 9 (2): CD007480. doi:10.1002/14651858.CD007480.pub3. PMC 6457829. PMID 27681486.
- ↑ "Vitamin A 25,000 Softgels". NOW Foods. Archived from the original on 23 December 2021. Retrieved 23 December 2021.
- 1 2 Institute of Medicine (US) Panel on Dietary Antioxidants Related Compounds (2000). "Beta-carotene and other Carotenoids". Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington, DC: The National Academies Press. pp. 325–82. doi:10.17226/9810. ISBN 978-0-309-06935-9. PMID 25077263. Archived from the original on 2 September 2017. Retrieved 19 December 2021.
- ↑ Maharshak N, Shapiro J, Trau H (March 2003). "Carotenoderma--a review of the current literature". Int J Dermatol. 42 (3): 178–81. doi:10.1046/j.1365-4362.2003.01657.x. PMID 12653910. S2CID 27934066.
- ↑ Nasser Y, Jamal Z, Albuteaey M (11 August 2021). "Carotenemia". StatPearls. doi:10.1007/s00253-001-0902-7. PMID 30521299. S2CID 22232461.
- ↑ "Federal Register May 27, 2016 Food Labeling: Revision of the Nutrition and Supplement Facts Labels" (PDF). Archived (PDF) from the original on 8 August 2016. Retrieved 8 June 2022.
- ↑ "Daily Value Reference of the Dietary Supplement Label Database (DSLD)". Dietary Supplement Label Database (DSLD). Archived from the original on 7 April 2020. Retrieved 18 December 2021.
- 1 2 3 "Rank order of vitamin A content in foods, retinol activity equivalent (RAE) in ug per 100 g". FoodData Central, US Department of Agriculture. 1 October 2021. Archived from the original on 3 April 2019. Retrieved 20 December 2021.
- ↑ "USDA National Nutrient Database for Standard Reference, Release 28" (PDF). 28 October 2015. Archived (PDF) from the original on 17 December 2021. Retrieved 5 February 2022.
- ↑ "Vitamin A 25,000 IU (7,500 μg)". WonderLabs. Archived from the original on 27 January 2022. Retrieved 26 January 2022.
- ↑ "Vital Nutrients Vitamin A 7,500 RAE". Pure Prescriptions. Archived from the original on 12 June 2022. Retrieved 26 January 2022.
- 1 2 3 4 "Total number of nutrients in food vehicles, according to a country's fortification standard". Global Fortification Data Exchange. Archived from the original on 11 April 2019. Retrieved 7 January 2022.
- ↑ FAQ: Who invented Golden Rice and how did the project start? Archived 14 February 2022 at the Wayback Machine Goldenrice.org.
- ↑ LSU AgCenter Communications (2004). "'Golden Rice' Could Help Reduce Malnutrition". Archived from the original on 28 September 2007.
- ↑ Kettenburg AJ, Hanspach J, Abson DJ, Fischer J (2018). "From disagreements to dialogue: unpacking the Golden Rice debate". Sustain Sci. 13 (5): 1469–82. doi:10.1007/s11625-018-0577-y. PMC 6132390. PMID 30220919.
- ↑ Ye X, Al-Babili S, Klöti A, Zhang J, Lucca P, Beyer P, Potrykus I (January 2000). "Engineering the provitamin A (beta-carotene) biosynthetic pathway into (carotenoid-free) rice endosperm". Science. 287 (5451): 303–5. Bibcode:2000Sci...287..303Y. doi:10.1126/science.287.5451.303. PMID 10634784.
- ↑ "Golden Rice meets food safety standards in three global leading regulatory agencies". International Rice Research Institute – IRRI. Archived from the original on 29 May 2018. Retrieved 30 May 2018.
- ↑ Talavera C. "Philippines OKs GMO 'golden rice'". Philstar.com. Archived from the original on 24 August 2021. Retrieved 21 August 2021.
- ↑ "Filipinos soon to plant and eat Golden Rice". Philippine Rice Research Institute. 23 July 2021. Archived from the original on 12 June 2022. Retrieved 21 August 2021.
- ↑ "Vitamin A supplementation coverage rate (children ages 6–59 months)". Our World in Data. Archived from the original on 23 April 2021. Retrieved 6 March 2020.
- ↑ Vitamin A Supplementation: A Decade of Progress (PDF). New York: UNICEF. 2007. p. 3. ISBN 978-92-806-4150-9. Archived from the original (PDF) on 31 October 2020. Retrieved 23 January 2011.
- 1 2 Wirth JP, Petry N, Tanumihardjo SA, Rogers LM, McLean E, Greig A, Garrett GS, Klemm RD, Rohner F (February 2017). "Vitamin A Supplementation Programs and Country-Level Evidence of Vitamin A Deficiency". Nutrients. 9 (3): 190. doi:10.3390/nu9030190. PMC 5372853. PMID 28245571.
- ↑ "Global prevalence of vitamin A deficiency in populations at risk 1995–2005" (PDF). WHO global database on vitamin A deficiency. World Health Organization. 2009. Archived (PDF) from the original on 15 December 2017. Retrieved 8 June 2022.
- ↑ Black RE, Allen LH, Bhutta ZA, Caulfield LE, de Onis M, Ezzati M, Mathers C, Rivera J (January 2008). "Maternal and child undernutrition: global and regional exposures and health consequences". The Lancet. 371 (9608): 243–260. doi:10.1016/S0140-6736(07)61690-0. PMID 18207566. S2CID 3910132. Archived from the original on 16 October 2021.
- ↑ Akhtar S, Ahmed A, Randhawa MA, Atukorala S, Arlappa N, Ismail T, Ali Z (December 2013). "Prevalence of vitamin A deficiency in South Asia: causes, outcomes, and possible remedies". Journal of Health, Population, and Nutrition. 31 (4): 413–423. doi:10.3329/jhpn.v31i4.19975. PMC 3905635. PMID 24592582.
- ↑ Tanumihardjo SA (August 2011). "Vitamin A: biomarkers of nutrition for development". The American Journal of Clinical Nutrition. 94 (2): 658S–665S. doi:10.3945/ajcn.110.005777. PMC 3142734. PMID 21715511.
- ↑ Saeed A, Dullaart RP, Schreuder TC, Blokzijl H, Faber KN (December 2017). "Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD)". Nutrients. 10 (1): 29. doi:10.3390/nu10010029. PMC 5793257. PMID 29286303.
- 1 2 Hensel P (2010). "Nutrition and skin diseases in veterinary medicine". Clin Dermatol. 28 (6): 686–93. doi:10.1016/j.clindermatol.2010.03.031. PMID 21034992.
- ↑ Clugston RD, Blaner WS (2014). "Vitamin A (retinoid) metabolism and actions: What we know and what we need to know about amphibians". Zoo Biol. 33 (6): 527–35. doi:10.1002/zoo.21140. PMC 4685470. PMID 24958673.
- ↑ Shojadoost B, Yitbarek A, Alizadeh M, Kulkarni RR, Astill J, Boodhoo N, Sharif S (April 2021). "Centennial Review: Effects of vitamins A, D, E, and C on the chicken immune system". Poult Sci. 100 (4): 100930. doi:10.1016/j.psj.2020.12.027. PMC 7900602. PMID 33607314.
- ↑ Semba RD (April 1999). "Vitamin A as "anti-infective" therapy, 1920-1940". J Nutr. 129 (4): 783–91. doi:10.1093/jn/129.4.783. PMID 10203551.
- ↑ "STANDARD FOR FISH OILS CODEX STAN 329-2017" (PDF). CODEX ALIMENTARIUS COMMISSION, World Health Organization. 2017. Archived (PDF) from the original on 3 March 2022. Retrieved 28 January 2022.
- ↑ André A, Ruivo R, Gesto M, Castro LF, Santos MM (November 2014). "Retinoid metabolism in invertebrates: when evolution meets endocrine disruption". Gen Comp Endocrinol. 208: 134–45. doi:10.1016/j.ygcen.2014.08.005. PMID 25132059.
- ↑ Gutierrez-Mazariegos J, Schubert M, Laudet V (2014). "Evolution of retinoic acid receptors and retinoic acid signaling". Subcell Biochem. Subcellular Biochemistry. 70: 55–73. doi:10.1007/978-94-017-9050-5_4. ISBN 978-94-017-9049-9. PMID 24962881.
- ↑ Albalat R (December 2009). "The retinoic acid machinery in invertebrates: ancestral elements and vertebrate innovations". Mol Cell Endocrinol. 313 (1–2): 23–35. doi:10.1016/j.mce.2009.08.029. PMID 19737598. S2CID 41188476.
- ↑ "Micronutrient Deficiencies-Vitamin A". World Health Organization. Archived from the original on 27 January 2011. Retrieved 9 April 2008.
- 1 2 Nickle SB, Peterson N, Peterson M (April 2014). "Updated Physician's Guide to the Off-label Uses of Oral Isotretinoin". J Clin Aesthet Dermatol. 7 (4): 22–34. PMC 3990537. PMID 24765227.
- ↑ "Tretinoin". The American Society of Health-System Pharmacists. 22 March 2021. Archived from the original on 30 November 2016. Retrieved 23 December 2021.
- ↑ Merritt B, Burkhart CN, Morrell DS (June 2009). "Use of isotretinoin for acne vulgaris". Pediatric Annals. 38 (6): 311–20. doi:10.3928/00904481-20090512-01. PMID 19588674.
- ↑ Layton A (May 2009). "The use of isotretinoin in acne". Dermato-Endocrinology. 1 (3): 162–9. doi:10.4161/derm.1.3.9364. PMC 2835909. PMID 20436884.
- ↑ Leyden J, Stein-Gold L, Weiss J (September 2017). "Why Topical Retinoids Are Mainstay of Therapy for Acne". Dermatol Ther (Heidelb). 7 (3): 293–304. doi:10.1007/s13555-017-0185-2. PMC 5574737. PMID 28585191.
- 1 2 Kang S, Voorhees JJ. Topical retinoids. In: Fitzpatrick's Dermatology in General Medicine, 7th ed, Wolff K, Goldsmith LA, Katz SI, et al (Eds), McGraw Hill, New York 2008. p.2106.
- ↑ Fernandez [Graber] EM, Zaenglein A, Thiboutot D. Acne Treatment Methodologies. In: Cosmetic Formulation of Skin Care Products, Taylor and Francis Group, New York 2006. p.273.
- ↑ Kovitwanichkanont T, Driscoll T (September 2018). "A comparative review of the isotretinoin pregnancy risk management programs across four continents". Int J Dermatol. 57 (9): 1035–1046. doi:10.1111/ijd.13950. PMID 29508918. S2CID 3726217.
- ↑ Scott LJ (November 2019). "Trifarotene: First Approval". Drugs. 79 (17): 1905–09. doi:10.1007/s40265-019-01218-6. PMID 31713811. S2CID 207964653.
- ↑ Mukherjee S, Date A, Patravale V, Korting HC, Roeder A, Weindl G (2006). "Retinoids in the treatment of skin aging: an overview of clinical efficacy and safety". Clinical Interventions in Aging. 1 (4): 327–48. doi:10.2147/ciia.2006.1.4.327. PMC 2699641. PMID 18046911.
- ↑ Boehnlein J, Sakr A, Lichtin JL, Bronaugh RL (1994). "Characterization of Esterase and Alcohol Dehydrogenase Activity in Skin. Metabolism of Retinyl Palmitate to Retinol (Vitamin A) During Percutaneous Absorption". Pharmaceutical Research. 11 (8): 1155–59. doi:10.1023/A:1018941016563. ISSN 0724-8741. PMID 7971717. S2CID 25458156.
- ↑ Villani A, Annunziata MC, Cinelli E, Donnarumma M, Milani M, Fabbrocini G (October 2020). "Efficacy and safety of a new topical gel formulation containing retinol encapsulated in glycospheres and hydroxypinacolone retinoate, an antimicrobial peptide, salicylic acid, glycolic acid and niacinamide for the treatment of mild acne: preliminary results of a 2-month prospective study". G Ital Dermatol Venereol. 155 (5): 676–79. doi:10.23736/S0392-0488.20.06581-5. PMID 32869963. S2CID 221402891.
- ↑ Sun L, Atkinson CA, Lee YG, Jin YS (November 2020). "High-level β-carotene production from xylose by engineered Saccharomyces cerevisiae without overexpression of a truncated HMG1 (tHMG1)". Biotechnol Bioeng. 117 (11): 3522–32. doi:10.1002/bit.27508. OSTI 1651205. PMID 33616900. S2CID 222411819.
- ↑ Wittig G.; Pommer H.: DBP 954247, 1956
- ↑ Wittig G.; Pommer H. (1959). Chem. Abstr. 53: 2279
- ↑ US 2609396, Inhoffen Hans Herloff & Pommer Horst, "Compounds with the carbon skeleton of beta-carotene and process for the manufacture thereof", published 1952-09-02
- 1 2 3 Parker GL, Smith LK, Baxendale IR (February 2016). "Development of the industrial synthesis of vitamin A". Tetrahedron. 72 (13): 1645–52. doi:10.1016/j.tet.2016.02.029.
- ↑ Dow Jones Business News (21 November 2001). "Vitamin Makers Fined Record $755.1 Million in Price-Fixing Case". The New York Times. Archived from the original on 24 March 2022. Retrieved 24 March 2022.
- ↑ Sun L, Kwak S, Jin YS (September 2019). "Vitamin A Production by Engineered Saccharomyces cerevisiae from Xylose via Two-Phase in Situ Extraction". ACS Synth Biol. 8 (9): 2131–40. doi:10.1021/acssynbio.9b00217. OSTI 1547320. PMID 31374167. S2CID 199389319.
- ↑ Druesne-Pecollo N, Latino-Martel P, Norat T, Barrandon E, Bertrais S, Galan P, Hercberg S (July 2010). "Beta-carotene supplementation and cancer risk: a systematic review and metaanalysis of randomized controlled trials". International Journal of Cancer. 127 (1): 172–84. doi:10.1002/ijc.25008. PMID 19876916. S2CID 24850769.
- ↑ He J, Gu Y, Zhang S (December 2018). "Vitamin A and Breast Cancer Survival: A Systematic Review and Meta-analysis". Clin Breast Cancer. 18 (6): e1389–e1400. doi:10.1016/j.clbc.2018.07.025. PMID 30190194. S2CID 52169953.
- ↑ Leelakanok N, D'Cunha RR, Sutamtewagul G, Schweizer ML (June 2018). "A systematic review and meta-analysis of the association between vitamin A intake, serum vitamin A, and risk of liver cancer". Nutr Health. 24 (2): 121–31. doi:10.1177/0260106018777170. PMID 29792083. S2CID 43944154.
- ↑ Tang JE, Wang RJ, Zhong H, Yu B, Chen Y (April 2014). "Vitamin A and risk of bladder cancer: a meta-analysis of epidemiological studies". World J Surg Oncol. 12: 130. doi:10.1186/1477-7819-12-130. PMC 4030017. PMID 24773914.
- ↑ Liu Y, Yu Q, Zhu Z, Zhang J, Chen M, Tang P, Li K (January 2015). "Vitamin and multiple-vitamin supplement intake and incidence of colorectal cancer: a meta-analysis of cohort studies". Med Oncol. 32 (1): 434. doi:10.1007/s12032-014-0434-5. PMID 25491145. S2CID 25904916.
- 1 2 Xu X, Yu E, Liu L, Zhang W, Wei X, Gao X, Song N, Fu C (November 2013). "Dietary intake of vitamins A, C, and E and the risk of colorectal adenoma: a meta-analysis of observational studies". Eur J Cancer Prev. 22 (6): 529–39. doi:10.1097/CEJ.0b013e328364f1eb. PMID 24064545. S2CID 36958552.
- ↑ Li K, Zhang B (August 2020). "The association of dietary β-carotene and vitamin A intake on the risk of esophageal cancer: a meta-analysis". Rev Esp Enferm Dig. 112 (8): 620–26. doi:10.17235/reed.2020.6699/2019. PMID 32543872. S2CID 219724574.
- ↑ Wu Y, Ye Y, Shi Y, Li P, Xu J, Chen K, Xu E, Yang J (August 2015). "Association between vitamin A, retinol intake and blood retinol level and gastric cancer risk: A meta-analysis". Clin Nutr. 34 (4): 620–6. doi:10.1016/j.clnu.2014.06.007. PMID 25008141.
- ↑ Wang Q, He C (April 2020). "Dietary vitamin A intake and the risk of ovarian cancer: a meta-analysis". Biosci Rep. 40 (4). doi:10.1042/BSR20193979. PMC 7138903. PMID 32149329.
- ↑ Zhang T, Chen H, Qin S, Wang M, Wang X, Zhang X, Liu F, Zhang S (December 2016). "The association between dietary vitamin A intake and pancreatic cancer risk: a meta-analysis of 11 studies". Biosci Rep. 36 (6). doi:10.1042/BSR20160341. PMC 5293573. PMID 27756825.
- 1 2 Yu N, Su X, Wang Z, Dai B, Kang J (November 2015). "Association of Dietary Vitamin A and β-Carotene Intake with the Risk of Lung Cancer: A Meta-Analysis of 19 Publications". Nutrients. 7 (11): 9309–24. doi:10.3390/nu7115463. PMC 4663591. PMID 26569298.
- ↑ Zhang YP, Chu RX, Liu H (2014). "Vitamin A intake and risk of melanoma: a meta-analysis". PLOS ONE. 9 (7): e102527. Bibcode:2014PLoSO...9j2527Z. doi:10.1371/journal.pone.0102527. PMC 4105469. PMID 25048246.
- ↑ Zhang X, Dai B, Zhang B, Wang Z (February 2012). "Vitamin A and risk of cervical cancer: a meta-analysis". Gynecol Oncol. 124 (2): 366–73. doi:10.1016/j.ygyno.2011.10.012. PMID 22005522.
- ↑ Kong P, Cai Q, Geng Q, Wang J, Lan Y, Zhan Y, Xu D (2014). "Vitamin intake reduce the risk of gastric cancer: meta-analysis and systematic review of randomized and observational studies". PLOS ONE. 9 (12): e116060. Bibcode:2014PLoSO...9k6060K. doi:10.1371/journal.pone.0116060. PMC 4280145. PMID 25549091.
- 1 2 Fainsod A, Bendelac-Kapon L, Shabtai Y (2020). "Fetal Alcohol Spectrum Disorder: Embryogenesis Under Reduced Retinoic Acid Signaling Conditions". Subcell Biochem. Subcellular Biochemistry. 95: 197–225. doi:10.1007/978-3-030-42282-0_8. ISBN 978-3-030-42280-6. PMID 32297301. S2CID 215793789.
- 1 2 Petrelli B, Bendelac L, Hicks GG, Fainsod A (January 2019). "Insights into retinoic acid deficiency and the induction of craniofacial malformations and microcephaly in fetal alcohol spectrum disorder". Genesis. 57 (1): e23278. doi:10.1002/dvg.23278. PMID 30614633. S2CID 58603210.
- ↑ "Fetal Alcohol Exposure". April 2015. Archived from the original on 10 June 2015. Retrieved 16 December 2021.
- ↑ Farrés J, Moreno A, Crosas B, Peralba JM, Allali-Hassani A, Hjelmqvist L, et al. (September 1994). "Alcohol dehydrogenase of class IV (sigma sigma-ADH) from human stomach. cDNA sequence and structure/function relationships". European Journal of Biochemistry. 224 (2): 549–557. doi:10.1111/j.1432-1033.1994.00549.x. PMID 7925371.
- ↑ Edenberg HJ, McClintick JN (December 2018). "Alcohol Dehydrogenases, Aldehyde Dehydrogenases, and Alcohol Use Disorders: A Critical Review". Alcoholism, Clinical and Experimental Research. 42 (12): 2281–2297. doi:10.1111/acer.13904. PMC 6286250. PMID 30320893.
- ↑ Shabtai Y, Fainsod A (April 2018). "Competition between ethanol clearance and retinoic acid biosynthesis in the induction of fetal alcohol syndrome". Biochem Cell Biol. 96 (2): 148–160. doi:10.1139/bcb-2017-0132. PMID 28982012.
- ↑ Shabtai Y, Bendelac L, Jubran H, Hirschberg J, Fainsod A (January 2018). "Acetaldehyde inhibits retinoic acid biosynthesis to mediate alcohol teratogenicity". Sci Rep. 8 (1): 347. Bibcode:2018NatSR...8..347S. doi:10.1038/s41598-017-18719-7. PMC 5762763. PMID 29321611.
- 1 2 3 Sanjoaquin MA, Molyneux ME (June 2009). "Malaria and vitamin A deficiency in African children: a vicious circle?". Malar J. 8: 134. doi:10.1186/1475-2875-8-134. PMC 2702350. PMID 19534807.
- 1 2 Semba RD (2012). "On the 'discovery' of vitamin A". Annals of Nutrition & Metabolism. 61 (3): 192–198. doi:10.1159/000343124. PMID 23183288. S2CID 27542506.
- ↑ Rosenfeld L (April 1997). "Vitamine—vitamin. The early years of discovery". Clinical Chemistry. 43 (4): 680–685. doi:10.1093/clinchem/43.4.680. PMID 9105273.
- ↑ Arens JF, Van Dorp DA (February 1946). "Synthesis of some compounds possessing vitamin A activity". Nature. 157 (3981): 190–191. Bibcode:1946Natur.157..190A. doi:10.1038/157190a0. PMID 21015124. S2CID 27157783.
- ↑ Van Dorp DA, Arens JF (August 1947). "Synthesis of vitamin A aldehyde". Nature. 159 (4058): 189. Bibcode:1947Natur.160..189V. doi:10.1038/160189a0. PMID 20256189. S2CID 4137483.
- ↑ Smith, K. Annabelle (13 August 2013). "A WWII Propaganda Campaign Popularized the Myth That Carrots Help You See in the Dark". Smithsonian.com. Archived from the original on 15 April 2021. Retrieved 2 May 2018.
- ↑ "The Nobel Prize in Physiology or Medicine 1967". Nobel Foundation. Archived from the original on 4 December 2013. Retrieved 28 July 2007.
- ↑ Ebrey T, Koutalos Y (January 2001). "Vertebrate photoreceptors". Progress in Retinal and Eye Research. 20 (1): 49–94. doi:10.1016/S1350-9462(00)00014-8. PMID 11070368. S2CID 2789591.
- ↑ Fridericia LS, Holm E (1925). "Experimental contribution to the study of the relation between night blindness and malnutrition". Am J Physiol. 73: 63–78. doi:10.1152/ajplegacy.1925.73.1.63.
External links
| Identifiers: |
|
|---|
- Vitamin A at the US National Library of Medicine Medical Subject Headings (MeSH)
- WHO publications on Vitamin A Deficiency