Aromatase deficiency
| Aromatase deficiency | |
|---|---|
| Other names: Congenital estrogen deficiency[1] | |
 | |
| AES results when the function of aromatase is impaired. The aromatase protein (pictured) is required for the biosynthesis of oestrogens like oestradiol in the human body. | |
| Specialty | Endocrinology |
| Complications | Virilisation, tall stature, primary amenorrhea, multicystic ovaries, |
| Usual onset | Adulthood |
| Duration | Lifetime |
| Types | Endocrine Disruptive Disorder |
| Causes | Genetic mutations of CPY19 |
| Diagnostic method | Extremely low level of oestrogen and elevated level of androgens |
| Treatment | Transdermal oestradiol replacement, hormone replacement therapy |
Aromatase deficiency is an exceedingly rare condition characterized by extremely low levels or complete absence of the enzyme aromatase activity in the body.[2] It is an autosomal recessive disease resulting from various mutations of gene CPY19 (P450arom) which can lead to delayed puberty in females, osteoporosis in males and virilization in pregnant mothers. As of 2016, only 35 cases have been described in medical literature.[3]
Signs and symptoms
The deficiency causes the virilization of XX fetuses. The onset of symptoms usually occurs in adolescence or early adulthood. The lack of estrogen results in the presentation of primary amenorrhea and tall stature. The taller than expected height occurs because estrogen normally causes fusion of the epiphyseal growth plates in the bones, and in its absence, the patient will keep growing longer. The gonadotropins LH and FSH will both be elevated and patients present with polycystic ovaries. Furthermore, the low oestrogen will predispose those with the condition to osteoporosis.[2]
Female
- After birth, female infants usually display ambiguous genitalia including labioscrotal fusion, clitoromegaly, and phallic genitalia. Hyperandrogenism is present at birth along with low level of estrogen in the blood. However, they have normal internal female genitalia.[4] Other presented symptoms assist with the correct diagnostics.
- During pubertal age, progressive sign of virilization such as growing of body hair can be observed along with puberty failure due to the lack of estradiol action.[2] The disruption of the LHRH-LH/FSH axis causes bone age to delay with the absence of growth spurt.
- In adulthood, symptoms include virilization, absence of breast development, primary amenorrhea and infertility, and multicystic ovaries.[4]
- Other symptoms include hypergonadotropic hypogonadism, polycystic ovaries, hypoplastic ovaries and tall stature.[3]
Male
Symptoms are generally manifested in adulthood:
- Tall stature, osteopenia, osteoporosis, Type II Diabetes, hyperinsulinemia, acanthusis nigricans, lipid metabolism disorders and liver function impairment.[4]
During pregnancy
During gestation, a baby with Aromatase Deficiency can cause a mother to become virilized by causing the deepening of the voice, cystic acne, more hair growth than normal, cliteromegaly, and hirsutism.[3] The mother also has an increased level of circulating testosterone.[5] However, the symptoms normally regress post-partum.[2]
Comorbidity
Aromatase deficiency may be comorbid with Autism through their mutual relationship with RORA deficiency. This affects both males and females however the effect on males is more common due to the female protective effect. RORA is the gene for aromatase, an enzyme that converts male to female hormones. Thus, RORA deficiency is linked to aromatase deficiency, which in turn can lead to elevated testosterone levels, a proposed risk factor for autism.[6]
Complications
Pregnant mother
Aromatase is an estrogen synthase that synthesize estrone (E1) and estradiol (E2) from Androstenedione and Testosterone respectively.[7] During pregnancy, the placenta, which is fetal tissue, synthesizes large amounts of the intermediates in the biosynthesis of the estrogens, androstenedione and testosterone, but cannot convert them to estrogens due to the absence of aromatase.[7] The levels of accumulated androgens in the mother can elevate 100-fold higher than normal cycling levels which subsequently virilise both the mother and the fetus. The mother will experience cystic acne, deepening of the voice and hirsutism.[2] However, these symptoms are normally resolved following parturition.[2]
If the fetus is a male, it will develop a normal male genitalia and will proceed to grow normally and exhibit secondary male sex characteristics.[8] If the fetus is a female, it will be born with ambiguous genitalia including labioscrotal fusion and a greatly enlarged phallus.[7]
Female
Aromatase deficient female cannot synthesize estrone or estradiol in the absence of aromatase. The amount of androgen will accumulate at a very high rate in the blood, disrupting the LHRH-LH/FSH axis that can potentially lead to polycystic ovaries in adulthood.[4] In the absence of estrogen, high level of circulating LH and FSH can results in Hypergonadotropic hypogonadism.[9]
While females begin to virilise and grow hair in various places during adolescent, they are unable to menstruate without the presence of estradiol, subsequently causing primary amenorrhea, clitormegaly, and absence of breast development.[2] As puberty fails, the growth spurt is absence and bone age is delayed.[4] Without treatment, the collection of excessive androgen in the blood can lead to development of polycystic ovaries.[2]
Male
Aromatase deficient males experience a normal growth into adulthood. With a very low level of circulating estrogen (<7pg/mL), resulting in a higher level of FSH and LH in the blood.[2] Elevated level of androgens do not contribute to harmonic skeletal muscle growth like estrogen, thus, patients exhibits eunuchoid body habitus.[4]
Patients are generally tall in stature and have a pattern of persistent linear bone growth into adulthood.[2][7] Without estrogen, the epiphyseal plates cannot fuse together properly, resulting in continuous height growth. As a necessary steroid to maintain bone homeostasis, low level of estrogen also result osteopenia and osteoporosis of the lumbar spine and cortical bone.[2][4] Estrogen is also thought to be linked to the abnormal lipid profile and hyperinsulinemia in men, however, the detail mechanism is unknown.[2]
Cause
Gene Mutation

Aromatase deficiency is an autosomal recessive disease with most of the mutations occur along the highly conservative regions of the gene. Both homozygous and heterozygous mutations have been identified along various location of the exon on the P450 arom (CYP19) gene localized on chromosome15p21.1.[8] In addition, mutations in cytochrome P450 oxidoreductase (POR), which is required for enzymatic activity of aromatase, can also cause aromatase deficiency.[10]
| Gender | Mutation | Transcription Results | Aromatase Activity (%) |
|---|---|---|---|
| Female | GT to GC at the 5’ Terminus of intron VI | An extra 87 bp insertion, between exon VI and intron VI | 0.3% |
| Female/Male | Single base change at bp 1123: C to T in exon X | Cysteine being transcribed instead of Arginine at position 375 (R375C) | 0.2% |
| Female | Point mutation (R457X) in exon X | No Transcription | - |
| Female | Mutation Valine 370 to Methionine in exon IX | - | - |
| Female | 1600 bp deletion in exon V | Aromatase lacking 59 Amino Acids | - |
| Female | Point mutation in exon X (R435C) | Missense mutation that causes loss of function | - |
| Female | Deletion of a single Phenylalanine residue at codon 234 in exon VI | - | - |
| Female | 568C insertion in CYP19A1 | 190 Leucine was changed to Proline | - |
| Female | Single base change at bp 1094 (G to A) in exon IX | Glutamine instead of Arginine being transcribed at position 365 (R365Q) | 0.4 |
| Male | C-base deletion in exon V | Resulting in a stop codon after 21 codons | 0.0 |
| Male | C to A substitution in intron V, at 3’ splicing acceptor site before exon VI | Premature stop codon | - |
| Male | Insertion of 21 bp at the codon 353 in exon IX | - | - |
| Male | Single base change at bp 628 (G to A) in the last nucleotide of exon V | Glutamic acid instead of a Lysine being transcribed at position 210 (E210K) | 1.0 |
| Gender | Mutation | Transcription Results | Aromatase Activity (%) |
|---|---|---|---|
| Female | Single base changes in exon X at bp 1303: C to T | Cysteine was transcribed instead of Arginine at position 435 (R435C) | 1.1 |
| Single base changes in exon X at bp 1310: G to A | Tyrosine was transcribed instead of Cysteine at position 437 (C437Y) | 0.0 | |
| Female | Point mutation (G to A) at the splicing point between exon and intron III | No transcription | 0.0 |
| Base pair deletion occurring at P408 (CCC) in exon IX | Nonsense codon 111 bp were transcribed down in the CYP19 | 0.0 | |
| Female | Point mutation (GAA to AAA) at bp 628 in exon V | Glutamic acid transcribed instead of lysine at position 210 (E210K) | 0.0 |
| A Base pair deletion occurring at E412 in exon IX | Transcribed a stop codon 98 bp downstream | 0.0 | |
| Male | Point mutation (ATG to AGG) at bp 380 in exon IV | Methionine was transcribed instead of arginine at position 127 (M127R) | - |
| Point mutation (CGC to CAC) at bp 1123 in exon IX | 2. Arginine was transcribed instead of histidine at position 375 (R375H) | - | |
| Male | 23 bp deletion in exon IV | Premature stop codon in exon IV | - |
| Point mutation (G to T) at first bp in intron IX | Alternative splicing? | - | |
Diagnosis
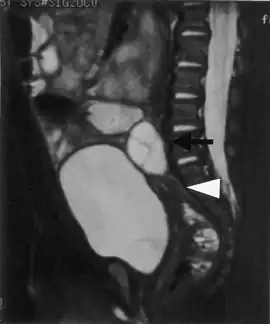
A fetus can be predicted to be suffering from aromatase deficiency when its pregnant mother is displaying virilization. A female infant can be physically diagnosed due to the abnormal genitalia along with hormonal blood test.[4] Excessively low level of estrogen and elevated level of androgens are diagnostic markers for aromatase deficiency in both males and females.[9] Testosterone level in the urine may be normal or elevated.[4]
Treatment
In males, transdermal estradiol replacement enable epiphyseal plates closure, increases bone density, promote skeletal maturation, lower FSH and LH level to normal and decrease insulin blood concentration.[4] In a young man with high stature due to unfused epiphysis, estrogen patch treatment daily possibly for life resolved the issue with further growth and osteoporosis.
In females, hormonal replacement therapy such as cyclic oral therapy of conjugated estrogen leads to breast development, menses, pubertal growth spurt, resolution of ovarian cysts, suppression of elevated FSH and LH levels in the blood, and proper bone growth.[4] Ambiguous genitalia, clitoromegaly, and ovarian cysts can be removed surgically[2] (forasmuch as not illegal).
History
Aromatase deficiency was first recorded in literature in 1991 by Shouz and colleagues.[11] The pregnant mother had low estrogen serum level and high androgens level in the third trimester along with signs of progressive virilisation. Upon delivery, the female infant exhibited pseudohermaphroditism. Aromatase activity of the placenta was approximately ten times less than the normal range.
See also
References
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Aromatase deficiency". www.orpha.net. Archived from the original on 28 November 2017. Retrieved 14 April 2019.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K (December 1995). "Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of oestrogens". J. Clin. Endocrinol. Metab. 80 (12): 3689–98. doi:10.1210/jcem.80.12.8530621. PMID 8530621.
- 1 2 3 Akçurin S, Türkkahraman D, Kim WY, Durmaz E, Shin JG, and Lee SJ (2016). "HA novel null mutation in P450 aromatase gene (CYP19A1) associated with development of hypoplastic ovaries in humans". J Clin Res Pediatr Endocrinol. 8 (2): 205–10. doi:10.4274/jcrpe.2761. PMC 5096477. PMID 27086564.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Zirilli L, Rochira V, Diazzi C, Caffagni G, Carani C (April 2008). "Human models of aromatase deficiency". J. Steroid Biochem. Mol. Biol. 109 (3–5): 212–8. doi:10.1016/j.jsbmb.2008.03.026. PMID 18448329. S2CID 23048215.
- ↑ Jones ME, Boon WC, McInnes K, Maffei L, Carani C, Simpson ER (May 2007). "Recognizing rare disorders: aromatase deficiency". Nat Clin Pract Endocrinol Metab. 3 (5): 414–21. doi:10.1038/ncpendmet0477. PMID 17452968. S2CID 8581107.
- ↑ Valerie W Hu et al. Investigation of sex differences in the expression of RORA and its transcriptional targets in the brain as a potential contributor to the sex bias in autism. Molecular Autism, May 2015 DOI: 10.1186/2040-2392-6-7
- 1 2 3 4 Blakemore J, Naftolin F (July 2016). "Aromatase: Contributions to Physiology and Disease in Women and Men". Physiology. 31 (4): 258–269. doi:10.1152/physiol.00054.2015. PMID 27252161.
- 1 2 Bulun, Serdar E. (2014). "Aromatase and estrogen receptor α deficiency". Fertility and Sterility. 101 (2): 323–329. doi:10.1016/j.fertnstert.2013.12.022. ISSN 0015-0282. PMC 3939057. PMID 24485503.
- 1 2 3 Mazen I, Mcelreavey K, Elaidy A, Kamel AK, Abdel-Hamid MS (January 2018). "Aromatase deficiency due to homozygous CYP19A1 mutation in a 46, XX Egyptian patient with ambiguous genitalia". Sex Dev. 11 (5–6): 275–279. doi:10.1159/000485278. PMID 29324451. S2CID 3595100.
- ↑ Parween, Shaheena; Fernández-Cancio, Mónica; Benito-Sanz, Sara; Camats, Núria; Velazquez, Maria Natalia Rojas; López-Siguero, Juan-Pedro; Udhane, Sameer S.; Kagawa, Norio; Flück, Christa E.; Audí, Laura; Pandey, Amit V. (2020). "Molecular basis of CYP19A1 deficiency in a 46, XX patient with R550W mutation in POR: Expanding the PORD phenotype". The Journal of Clinical Endocrinology & Metabolism. 105 (4): e1272–e1290. doi:10.1210/clinem/dgaa076. PMID 32060549.
- ↑ Shouzu M, Akasofu K, Harada T, Kubota Y (March 1991). "A new cause of female pseudohermaphroditism: placental aromatase deficiency". J. Clin. Endocrinol. Metab. 72 (3): 560–566. doi:10.1210/jcem-72-3-560. PMID 1825497.
Further reading
- Bulun SE (2014). "Aromatase and estrogen receptor α deficiency". Fertil. Steril. 101 (2): 323–9. doi:10.1016/j.fertnstert.2013.12.022. PMC 3939057. PMID 24485503.
External links
| Classification | |
|---|---|
| External resources |
|