Juvenile hemochromatosis
| Juvenile hemochromatosis | |
|---|---|
| Other names: Hemochromatosis type 2 | |
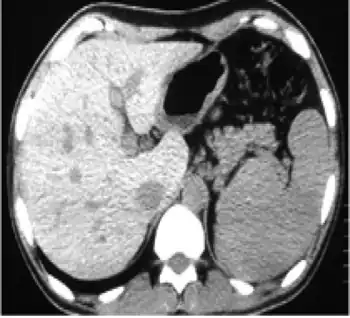 | |
| In non-contrast abdominal computed tomography, increased density of liver compared with spleen was reported | |
Juvenile hemochromatosis, also known as hemochromatosis type 2, is a rare form of hereditary hemochromatosis, which emerges in young individuals, typically between 15 and 30 years of age, but occasionally later.[1][2] It is characterized by an inability to control how much iron is absorbed by the body, in turn leading to iron overload, where excess iron accumulates in many areas of the body and causes damage to the places it accumulates.[1][3]
It is a genetic disorder that can be caused by mutations in either the HJV (also called HFE2) or HAMP genes, and is inherited in an autosomal recessive fashion.[3] Depending on which of these genes is affected, the disease can be further subdivided into types 2A and 2B.[2]
Signs and Symptoms
The most common symptoms of juvenile hemochromatosis are as follows:[2][3][4]
- Weakness
- Lethargy
- Hyperpigmentation (darkening of the skin)
- Arthropathy (joint disease)
- Diabetes
- Heart disease (dilated cardiomyopathy). Complications of heart disease are the main cause of death in those with untreated hemochromatosis.
- Hypogonadism (reduced activity of the genitals), which may result in decreased libido and infertility
- Amenorrhea in females
- Erectile dysfunction in males
- Loss of appetite
- Increased risk of infection by certain bacteria including V. vulnificus.
Other common complications include:
Less common symptoms and complications include:
- Osteoporosis
- Hepatomegaly (liver enlargement)
- Liver cirrhosis
- Cardiac arrhythmias
- Hypothyroidism
- Adrenocortical insufficiency
Genetics
Juvenile hemochromatosis can be caused by inheriting two mutated copies (alleles), one from each parent, of the genes for the proteins hemojuvelin (HFE2/HJV) or hepcidin (HAMP), and the disease can be subdivided into hemochromatosis types 2A and 2B according to which gene/protein is affected.[2][3]
- Type 2A is caused by inheriting two mutated alleles, one from each parent, for the HJV (aka HFE2) gene, which encodes the protein hemojuvelin.[2] Hemojuvelin is responsible for the maintaining correct levels of the protein hepcidin, which regulates iron absorption in the blood.[5] Without functional hemojuvelin, hepcidin levels are reduced, and the amount of iron absorbed into the blood during digestion is unable to be halted.[5][6]
- Type 2B is caused by inheriting two mutated alleles, one from each parent, for the HAMP gene, which encodes the protein hepcidin.[2] Hepcidin is responsible for regulating absorption of iron from the small intestine to the blood during the digestion of food, such to prevent blood iron levels from becoming too high. A lack of functional hepcidin prevents the body from stopping iron absorption when it has already reached adequate levels.[6]
Type 2A is the most common form, accounting for roughly 9 out of every 10 cases of the disease.[2]
Diagnosis
An individual may be suspected to have this condition based on their medical history, physical exam findings, and blood tests, and confirmation of the diagnosis can be made with further testing, often with use of gene panels.
Differential Diagnosis
Juvenile hemochromatosis shares signs and symptoms with many other conditions including:[2][3][4]
- Other types of hereditary hemochromatosis
- Atransferrinemia
- Aceruloplasminemia
- African iron overload
- Neonatal hemochromatosis
- Transfusional iron overload and other forms of secondary hemochromatosis
- Porphyria cutanea tarda
Blood Testing
The presence of hemochromatosis may be discovered incidentally on blood testing, or a diagnosis suspected based on symptoms may be supported or ruled out by blood testing. Elevated serum ferritin, an indicator of blood iron levels, and transferrin saturation, which is involved with absorption of iron from the gut, are very common.[2][3]
- Transferrin saturation may approach or reach 100%, where a normal value would lie between 16% and 45%. If transferrin saturation is normal, juvenile hemochromatosis can be ruled out.[4]
- Serum ferritin may only be slightly elevated as the disease progresses, however may quickly reach in excess of 1000ng/mL.[4]
Genetic Testing
In patients suspected to have juvenile hemochromatosis, the diagnosis can be confirmed through genetic testing for specific genes:[4]
- A single gene study may be considered in those that demonstrate iron overload at a very young age, and specifically looks for mutations in either the HJV (HFE2) or HAMP genes. As HJV is more commonly associated with the disease, this may be checked first, and if no alterations are found, HAMP may be tested next.
- A multi-gene study may be considered to more effectively search for genetic causes of the patient's symptoms, and can include HJV, HAMP, and other genes associated with similar conditions like HFE.
Imaging
MRI may be utilized in order to assess the extent to which iron has been deposited in certain tissues and organs, however does not have significant weight in the diagnosis of the condition.[4]
Biopsy
Liver biopsy, or removal of a small piece of liver tissue for analysis, can be done to assess the extent of iron overload in the liver, however is considered not to have a significant weight in the diagnosis of the condition.[4]
Treatment
Treatment for juvenile hemochromatosis is similar to that for other forms of hemochromatosis and iron overload, and focuses on reducing the amount of iron in the body in order to prevent complications of iron overload.[2][3][4] However, if the disease is not discovered early enough, or if progress is not well controlled, further treatments may be aimed at the symptoms of organ damage which may develop.
Phlebotomy
Phlebotomy, the removal of blood from the body, is the main treatment for juvenile hemochromatosis. One unit of blood, the amount typically given during blood donation, is typically removed per session, and it is generally recommended that this be done once weekly until acceptable levels of iron are in the blood, which may take years.[4] After these levels are reached, phlebotomy will be continued, but less often than once weekly, perhaps every few months.[3]
Chelation Therapy
In the event that phlebotomy is not an appropriate option or is not enough on its own to reduce iron levels, chelation medications, those that bind and remove certain metals from the blood, may be utilized.[2][3][4] Examples of chelators specifically for iron include deferoxamine and deferasirox.
Dietary Modification
It is recommended that those with juvenile hemochromatosis refrain from eating iron supplements, vitamin C supplements, and uncooked/undercooked seafood and shellfish, and reduce or eliminate consumption of alcoholic beverages and red meat.[1][3][4]
Additional Treatments
If the disease is advanced enough, further treatments can be aimed at the complications of the disease, depending on which are present:[2][4]
- Diabetes may be treated with oral medications and/or insulin as indicated.
- Heart failure may require followup with cardiology specialist, as well as medical treatment with certain medications including ACE inhibitors and diuretics.
- Hypogonadism may require treatment with hormone replacement therapy, which has also been shown to reduce the likelihood of developing osteoporosis later on.
- Liver cirrhosis may require beta blockers, a kind of antihypertensive medication.
- Joint pain, called arthralgias, can be treated with NSAIDs, a type of pain medication available over-the-counter.
Epidemiology
The incidence of juvenile hemochromatosis in the general population remains unknown at this time, however it is very rare. It more commonly occurs in those of European descent, becoming apparent during the first to third decades of life, and affects males and females at similar rates.[2][3][4]
References
- 1 2 3 "Hereditary Hemochromatosis - Stanford Children's Health". www.stanfordchildrens.org. Archived from the original on 2022-07-10. Retrieved 2022-03-31.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 "Juvenile Hemochromatosis". NORD (National Organization for Rare Disorders). Archived from the original on 2022-03-02. Retrieved 2022-04-03.
- 1 2 3 4 5 6 7 8 9 10 11 "Hemochromatosis type 2 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2021-03-23. Retrieved 2022-03-31.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Piperno, Alberto; Bertola, Francesca; Bentivegna, Angela (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Juvenile Hemochromatosis", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301349, archived from the original on 2022-01-21, retrieved 2022-04-05
- 1 2 "HJV gene: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 2021-11-02. Retrieved 2022-04-05.
- 1 2 "HAMP gene: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 2022-04-05. Retrieved 2022-04-05.
External links
- GeneReview/NIH/UW entry on Juvenile Hereditary Hemochromatosis Archived 2022-01-21 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|