Fibrin
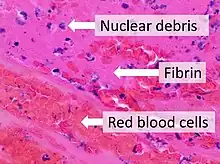

Fibrin (also called Factor Ia) is a fibrous, non-globular protein involved in the clotting of blood. It is formed by the action of the protease thrombin on fibrinogen, which causes it to polymerize. The polymerized fibrin, together with platelets, forms a hemostatic plug or clot over a wound site.
When the lining of a blood vessel is broken, platelets are attracted, forming a platelet plug. These platelets have thrombin receptors on their surfaces that bind serum thrombin molecules,[1] which in turn convert soluble fibrinogen in the serum into fibrin at the wound site. Fibrin forms long strands of tough insoluble protein that are bound to the platelets. Factor XIII completes the cross-linking of fibrin so that it hardens and contracts. The cross-linked fibrin forms a mesh atop the platelet plug that completes the clot.
Role in disease

Excessive generation of fibrin due to activation of the coagulation cascade leads to thrombosis, the blockage of a vessel by an agglutination of red blood cells, platelets, polymerized fibrin and other components. Ineffective generation or premature lysis of fibrin increases the likelihood of a hemorrhage.
Dysfunction or disease of the liver can lead to a decrease in the production of fibrin's inactive precursor, fibrinogen, or to the production of abnormal fibrinogen molecules with reduced activity (dysfibrinogenaemia). Hereditary abnormalities of fibrinogen (the gene is carried on chromosome 4) are both quantitative and qualitative in nature and include afibrinogenaemia, hypofibrinogenaemia, dysfibrinogenaemia, and hypodysfibrinogenemia.
Reduced, absent, or dysfunctional fibrin is likely to render patients as hemophiliacs.
Physiology

Fibrin from different animal sources is generally glycosylated with complex type biantennary asparagine-linked glycans. Variety is found in the degree of core fucosylation and in the type of sialic acid and galactose linkage.[2]
Structure

The image at the left is a crystal structure of the double-d fragment from human fibrin with two bound ligands. The experimental method used to obtain the image was X-ray diffraction, and it has a resolution of 2.30 Å. The structure is mainly made up of single alpha helices shown in red and beta sheets shown in yellow. The two blue structures are the bound ligands. The chemical structures of the ligands are Ca2+ ion, alpha-D-mannose (C6H12O6), and D-glucosamine (C6H13NO5).
See also
References
- ↑ Kehrel BE (2003). "[Blood platelets: biochemistry and physiology]". Hamostaseologie (in German). 23 (4): 149–58. doi:10.1055/s-0037-1619592. PMID 14603379.
- ↑ Pabst M, Bondili JS, Stadlmann J, Mach L, Altmann F (July 2007). "Mass + retention time = structure: a strategy for the analysis of N-glycans by carbon LC-ESI-MS and its application to fibrin N-glycans". Anal. Chem. 79 (13): 5051–7. doi:10.1021/ac070363i. PMID 17539604.
External links
- TGW1916.net, Defibrinated blood harvested from sheep (video)
- Fibrin: Molecule of the Month, by David Goodsell, RCSB Protein Data Bank
| Authority control: National libraries |
|---|