X-linked spinal muscular atrophy type 2
| X-linked spinal muscular atrophy type 2 | |
|---|---|
| Other names | Spinal muscular atrophy with arthrogryposis |
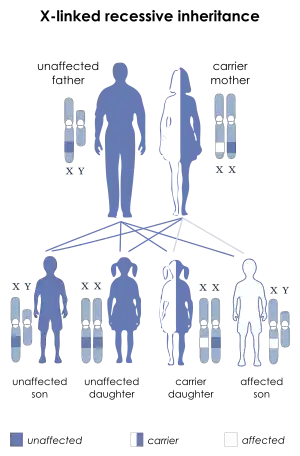 | |
| This condition is inherited in an X-linked recessive manner | |
| Specialty | Neurology |
X-linked spinal muscular atrophy type 2 (SMAX2, XLSMA), also known as arthrogryposis multiplex congenita X-linked type 1 (AMCX1), is a rare neurological disorder involving death of motor neurons in the anterior horn of spinal cord resulting in generalised muscle wasting (atrophy). The disease is caused by a mutation in UBA1 gene and is passed in an X-linked recessive manner by carrier mothers to affected sons.[1][2]
Affected babies have general muscle weakness, weak cry and floppy limbs; consequently, the condition is usually apparent at or even before birth. Symptoms resemble the more severe forms of the more common spinal muscular atrophy (SMA); however, SMAX2 is caused by a different genetic defect and only genetic testing can correctly identify the disease.
The disorder is usually fatal in infancy or early childhood due to progressive respiratory failure, although survival into teenage years has been reported.[3] As with many genetic disorders, there is no known cure to SMAX2. Appropriate palliative care may be able to increase quality of life and extend lifespan.
Signs and symptoms
XL-SMA is characterized by severe hypotonia and areflexia with loss of anterior horn cells in the spinal cord (i.e., lower motor neurons).[4] The disease course is similar to that in the most severe forms of classic autosomal recessive SMA caused by mutation of SMN1: SMA type 0 (SMA0) and SMA type I (SMA1).[4] In SMA0, prenatal onset of weakness and poor intrauterine movement results in congenital contractures.[4] In SMA1, motor skills regress before age six months; affected children are never able to sit independently.[4]
The weakness of XL-SMA is often prenatal in onset, manifests as polyhydramnios and poor movement in utero that results in congenital contractures.[4] Moreover, the weakness of XL-SMA is progressive.
Below is a list of known symptoms of XL-SMA:[5]
- Face :
- Myopathic Facies
- Facial weakness
- Mouth :
- Tongue fasciculations
- Respiratory :
- Insufficiency due to muscle weakness
- Chest :
- Chest deformities
- Genitourinary :
- External Genitalia (Male)
- Internal Genitalia (Male)
- Skeletal :
- Arthrogryposis
- Multiple joint contractures
- Bone fractures (at birth and postnatal)
- Skull-
- Dysmorphic Skull
- Hands-
- Digital contractures
- Skull-
Note: Clinical diagnosis of X-linked spinal muscular atrophy type 2 should be considered for children who exhibit the following:[4]
- Evidence of degeneration and loss of anterior horn cells in the spinal cord and brain stem
- Normal SMN1 molecular genetic testing to rule out autosomal recessive spinal muscular atrophy
- Male gender in a simplex case or X-linked pattern of inheritance in families with more than one affected individual
Genetics
X-linked spinal muscular atrophy type 2 is inherited in an X-linked recessive pattern. The gene associated with this disorder, UBA1, is located on the X chromosome at Xp11.3 and contains 27 exons; moreover, translation begins at the second exon.[6] In males, if the X chromosome contains an altered copy of the gene then the male will have the disorder. In females, a mutation of the gene on both X chromosomes would have to occur in order for the female to have the disorder. Since females are unlikely to have two altered copies of the gene, males are affected by this disorder much more frequently than females.
A recent study has resulted the in detection of three rare novel variants in exon 15 of UBA1 that segregated with the disease: two missense mutations present in each of one XLSMA family (314370.0001, 314370.0002), and one synonymous C-to-T substitution (314370.0003) identified in another three unrelated families.[6] Moreover, in a sixth family, neither of the two missense mutations or the synonymous substitution was identified.[6] Ramser et al. (2008) demonstrated that the synonymous C-to-T substitution leads to significant reduction of UBA1 expression and alters the methylation pattern of exon 15, implying a plausible role of this DNA element in developmental UBA1 expression in humans.[1][6] Thus, SMAX2 is one of several neurodegenerative disorders associated with defects in the ubiquitin-proteasome pathway.[6] The severity of SMAX2 does not waiver regardless of the type of causative mutation.
Diagnosis
Components that may lead to a diagnosis include the presence of clinical symptoms,[7] evidence of degeneration, and analysis of family history. One notable sign of SMAX2 is the loss of anterior horn cells in the spinal cord and brain stem. SMAX2 is typically confirmed through genetic testing that shows a mutation in the ubiquitin-like modifier-activating enzyme 1 gene (UBA1). The UBA1 gene is important to diagnosis as it is the only gene known to correspond with degeneration of XL-SMA. In infancy it is important to look for the following when considering a X-linked spinal muscular atrophy diagnosis:[8]
- Congenital hypotonia and areflexia on physical examination
- Congenital contractures/fractures
- Digital contractures at birth that remain throughout the child's life
- Evidence of degeneration and loss of anterior horn cells (i.e., lower motor neurons) in the spinal cord and brain stem
A child with these symptoms are likely to have X-linked spinal muscular atrophy. In order to confirm this the most practical measures to take next are:[8]
- to perform a normal SMN1 molecular genetic testing to rule out autosomal recessive spinal muscular atrophy
- perform an analysis to see if it is male gender in a simplex case (i.e., a single occurrence in a family) or find the presence of X-linked pattern of inheritance in families with more than one affected individual
Management
Following a diagnosis of the X-linked infantile spinal muscular atrophy it is recommended for the patient to go through further evaluations to alleviate symptoms as there is currently no known cure to SMAX2. The types of evaluations that are recommended are categorized in nutrition/feeding and respiratory function.
The nutrition and feeding evaluations are focused on determining if:[8]
- caloric intake is adequate
- dysphagia or fatigue while feeding occurs
- problems related to swallowing persist
- gastronomy tube placement and fundoplication are necessary
The respiratory evaluations are used to:[8]
- assess respiratory rate, work required to breath, signs of paradoxical breathing, the shape of the chest wall, and skin perfusion
- perform baseline pulmonary studies to establish the extent of restive airway disease and cough efficiency
- perform sleep evaluations to assess for sleep-disordered breathing
Other evaluations include assessing the existing contractures, neurologic evaluation to assess muscle tone and help provide supportive management, and highly recommended consultations with a genetic counselor.[8]
Overall treatment aims at alleviating the symptoms and may include mechanical ventilation, feeding tube, gastrostomy, and orthopedic interventions.
Epidemiology
X-linked spinal muscular atrophy type 2 is considered a rare disorder, and its prevalence is unknown.[9] Currently, only 14 multigenerational families with affected family members have been identified throughout North America, Europe, Mexico, and Thailand.[10]
See also
References
- 1 2 Ramser, J.; Ahearn, M. E.; Lenski, C.; Yariz, K. O.; Hellebrand, H.; Von Rhein, M.; Clark, R. D.; Schmutzler, R. K.; Lichtner, P.; Hoffman, E. P.; Meindl, A.; Baumbach-Reardon, L. (2008). "Rare Missense and Synonymous Variants in UBE1 Are Associated with X-Linked Infantile Spinal Muscular Atrophy". The American Journal of Human Genetics. 82 (1): 188–193. doi:10.1016/j.ajhg.2007.09.009. PMC 2253959. PMID 18179898.
- ↑ Dressman, D.; Ahearn, M. E.; Yariz, K. O.; Basterrecha, H.; Martínez, F.; Palau, F.; Barmada, M. M.; Clark, R. D.; Meindl, A.; Wirth, B.; Hoffman, E. P.; Baumbach-Reardon, L. (2007). "X-linked infantile spinal muscular atrophy: Clinical definition and molecular mapping". Genetics in Medicine. 9 (1): 52–60. doi:10.1097/GIM.0b013e31802d8353. PMID 17224690.
- ↑ Kobayashi, H.; Baumbach, L.; Matise, T. C.; Schiavi, A.; Greenberg, F.; Hoffman, E. (1995). "A gene for a severe lethal form of X-linked arthrogryposis (X-linked infantile spinal muscular atrophy) maps to human chromosome Xp11.3–q11.2". Human Molecular Genetics. 4 (7): 1213–1216. doi:10.1093/hmg/4.7.1213. PMID 8528211.
- 1 2 3 4 5 6 Baumbach-Reardon, Lisa; Sacharow, Stephanie J.; Ahearn, Mary Ellen (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Spinal Muscular Atrophy, X-Linked Infantile", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301739, retrieved 2021-05-03
- ↑ "OMIM Clinical Synopsis - #301830 - SPINAL MUSCULAR ATROPHY, X-LINKED 2; SMAX2". www.omim.org. Retrieved 2021-05-04.
- 1 2 3 4 5 "OMIM Entry - * 314370 - Ubiquitin-Like Modifier-Activating Enzyme 1; UBA1". OMIM. Retrieved 2021-05-04.
- ↑ "OMIM Clinical Synopsis - #301830 - SPINAL MUSCULAR ATROPHY, X-LINKED 2; SMAX2". www.omim.org. Retrieved 2021-05-04.
- 1 2 3 4 5 Baumbach-Reardon, Lisa; Sacharow, Stephanie J.; Ahearn, Mary Ellen (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Spinal Muscular Atrophy, X-Linked Infantile", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301739, retrieved 2021-05-03
- ↑ "X-linked infantile spinal muscular atrophy: MedlinePlus Genetics". medlineplus.gov. Retrieved 2021-05-03.
- ↑ Baumbach-Reardon, Lisa; Sacharow, Stephanie J.; Ahearn, Mary Ellen (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Spinal Muscular Atrophy, X-Linked Infantile", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301739, retrieved 2021-05-04
Further reading
- Dlamini, N.; Josifova, D. J.; Paine, S. M. L.; Wraige, E.; Pitt, M.; Murphy, A. J.; King, A.; Buk, S.; Smith, F.; Abbs, S.; Sewry, C.; Jacques, T. S.; Jungbluth, H. (2013). "Clinical and neuropathological features of X-linked spinal muscular atrophy (SMAX2) associated with a novel mutation in the UBA1 gene". Neuromuscular Disorders. 23 (5): 391–398. doi:10.1016/j.nmd.2013.02.001. PMID 23518311. S2CID 45706389.
- Baumbach-Reardon L.; Sacharow S.; Ahearn M. E. "Spinal Muscular Atrophy, X-Linked Infantile." 30 Oct 2008 [Updated 13 Sep 2012]. In: Pagon R. A.; Adam M. P.; Ardinger H. H.; et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2014. Available from: www
.ncbi ..nlm .nih .gov /books /NBK2594