Fazio–Londe disease
| Fazio–Londe disease | |
|---|---|
| Other names: Progressive bulbar palsy of childhood | |
 | |
| This condition has an autosomal recessive mode of inheritance | |
Fazio–Londe disease (FLD), also called progressive bulbar palsy of childhood,[1][2] is a very rare inherited motor neuron disease of children and young adults and is characterized by progressive paralysis of muscles innervated by cranial nerves.
Signs and symptoms
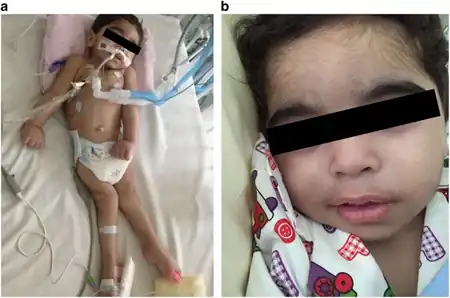
FLD produces rapidly progressive weakness of tongue, face and pharyngeal muscles in a clinical pattern similar to myasthenia. Neuromuscular transmission may be abnormal in these muscles because of rapid denervation and immature reinnervation. Paralysis occurs secondary to degeneration of the motor neurons of the brain stem. It causes progressive bulbar paralysis due to involvement of motor neurons of the cranial nerve nuclei. The most frequent symptoms at onset of progressive bulbar paralysis of childhood has been a unilateral facial paralysis. It is followed in frequency by dysarthria due to facial weakness or by dysphagia. Palatal weakness and palpebral ptosis also have been reported in few patients. Both sexes can be affected.[3][4]
Genetics
Fazio–Londe disease is linked to a genetic mutation in the SLC52A3 gene on chromosome 20 (locus: 20p13).[1] It is allelic and phenotypically similar to Brown–Vialetto–Van Laere syndrome.[1][5] The condition is inherited in an autosomal recessive manner.[1] The gene encodes the intestinal riboflavin transporter (hRFT2).
Diagnosis
Symptoms of Fazio-Londe include bulbar palsy, hearing loss, facial weakness, and difficulty breathing. The disease is caused by mutations in the SLC52A2 gene and the SLC52A1 (GPR172B) genes which code for hRFT3 and hRFT1, human riboflavin transporters. Only muscle biopsy and examination of the transporter genes is considered to provide a definitive diagnosis. However, because the disease is so often fatal without treatment, and because the treatment is so inexpensive and with little risk, it is recommended that if the disease is suspected that riboflavin therapy be started immediately while testing is in progress.[6]
Treatment
The condition is treatable.[7] High doses of oral riboflavin 5 phosphate may work.[6] If that didn't work sublingual FAD may work.
Prognosis
Onset of first symptom has been reported between 1–12 years, with a mean age of onset at 8 years. Clinical course can be divided into early (< 6 yrs age, predominance of respiratory symptoms) and late course (6–20 years of age, predominance of motor symptoms on superior limbs). Progression to involve other cranial nerve muscles occurs over a period of months or years. In the Gomez review facial nerve was affected in all cases while hypoglossal nerve was involved in all except one case. Other cranial nerves involved were vagus, trigeminal, spinal accessory nerve, abducens, oculomotor and glossopharyngeal in this order. Corticospinal tract signs were found in 2 of the 14 patients.
The disease may progress to patient's death in a period as short as 9 months or may have a slow evolution or may show plateaus. Postmortem examination of cases have found depletion of nerve cells in the nuclei of cranial nerves. The histologic alterations found in patient with Fazio–Londe disease were identical to those seen in infantile-onset spinal muscular atrophy.
Strength may improve with administration of cholinesterase inhibitors.
History
Berger, in 1876, first reported a case of 12-year-old child with progressive bulbar paralysis
Eponym
It is named for the Italian pathologist, Eugenio Fazio (1849–1902) and for the French physician, Paul Frederic Louis Londe (1864–1944).[8][9]
See also
References
- 1 2 3 4 Online Mendelian Inheritance in Man (OMIM): 211500
- ↑ McShane, MA; Boyd, S; Harding, B; Brett, EM; Wilson, J (December 1992). "Progressive bulbar paralysis of childhood. A reappraisal of Fazio-Londe disease". Brain: A Journal of Neurology. 115 (Pt 6): 1889–900. doi:10.1093/brain/115.6.1889. PMID 1486466.
- ↑ "OMIM Entry - # 211500 - FAZIO-LONDE DISEASE". www.omim.org. Archived from the original on 16 February 2020. Retrieved 18 July 2021.
- ↑ "Fazio-Londe Disease - Tests - GTR - NCBI". www.ncbi.nlm.nih.gov. Archived from the original on 11 August 2023. Retrieved 18 July 2021.
- ↑ Dipti S, Childs AM, Livingston JH, et al. (September 2005). "Brown–Vialetto–Van Laere syndrome; variability in age at onset and disease progression highlighting the phenotypic overlap with Fazio–Londe disease". Brain Dev. 27 (6): 443–446. doi:10.1016/j.braindev.2004.10.003. PMID 16122634. S2CID 32223440.
- 1 2 Bosch, Annet M; Stroek, Kevin; Abeling, Nico G; Waterham, Hans R; IJIst, Loedijk; Wanders, Ronald JA (2012). "The Brown-Vialetto-Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives". Orphanet Journal of Rare Diseases. BMC. 7: 83. doi:10.1186/1750-1172-7-83. PMC 3517535. PMID 23107375.
- ↑ Varadarajan, Poovazhagi; Thayanathi, Vimal; Pauline, LeemaC (2014). "Fazio Londe syndrome: A treatable disorder". Annals of Indian Academy of Neurology. 18 (1): 87–9. doi:10.4103/0972-2327.144283. PMC 4350224. PMID 25745320.
- ↑ synd/1909 at Who Named It?
- ↑ Londe, P. Paralysie bulbaire progressive, infantile et familiale. Rev. Med. 14: 212–254, 1894.
| Classification |
|---|