Johanson–Blizzard syndrome
| Johanson-Blizzard syndrome | |
|---|---|
| Other names: JBS | |
 | |
| Photographs of individuals with Johanson-Blizzard Syndrome showing characteristic facial features. | |
| Symptoms | pancreatic insufficiency, intellectual disability, distinctive craniofacial abnormalities, intestinal malabsorption, deafness, dental abnormalities |
| Differential diagnosis | cystic fibrosis, schwachman syndrome, Pearson marrow-pancreas syndrome |
Johanson–Blizzard syndrome is a rare, sometimes fatal autosomal recessive multisystem congenital disorder featuring abnormal development of the pancreas, nose and scalp, with intellectual disability, hearing loss and growth failure.[1] It is sometimes described as a form of ectodermal dysplasia.[2]
The disorder is especially noted for causing profound developmental errors and exocrine dysfunction of the pancreas, and it is considered to be an inherited pancreatic disease.[3]
Signs and Symptoms
Exocrine
The most prominent effect of Johanson–Blizzard syndrome is pancreatic exocrine insufficiency.[1][4][5][6][7] Varying degrees of decreased secretion of lipases, pancreatic juices such as trypsin, trypsinogen and others, as well as malabsorption of fats and disruptions of glucagon secretion and its response to hypoglycemia caused by insulin activity are major concerns when Johanson–Blizzard syndrome is diagnosed.[1][3][8] Associated with developmental errors, impaired apoptosis, and both prenatal and chronic inflammatory damage, necrosis and fibrosis of the pancreatic acini (clusters of pancreatic exocrine gland tissue, where secretion of pancreatic juice and related enzymes occurs), pancreatic exocrine insufficiency in Johanson–Blizzard syndrome can additionally stem from congenital replacement of the acini with fatty tissue.[1][3][8][9][10] Near total replacement of the entire pancreas with fatty tissue has also been reported. This is a progressive, sometimes fatal consequence of the disorder.[9]
Endocrine
Endocrine insufficiency of the pancreas occurs with Johanson–Blizzard syndrome, though it is sometimes less common and less pronounced than the more prominent effects on exocrine function.[1] The islets of Langerhans are ducts in the pancreas where endocrine activity such as the release of hormones glucagon, somatostatin and insulin takes place. Pancreatic endocrine insufficiency in Johanson–Blizzard syndrome can be associated with either a buildup of connective tissue in the islet regions, congenital replacement of the islets with fatty tissue, or improper nerve signalling to the islets.[1][5][8][11][12] Endocrine dysfunction of the pancreas often results in diabetes mellitus. Both insulin resistance and diabetes have been observed with Johanson–Blizzard syndrome, and it is suggested that diabetes should be considered as a complication of Johanson–Blizzard syndrome and its course.[5][11]
Ductular output of fluids and electrolytes is preserved in the pancreas of many with Johanson–Blizzard syndrome, as well as moderate to normal levels of functioning bicarbonate.[1]
Endocrine abnormalities in other areas have also been present with the disorder. These include hypothyroidism,[2] growth hormone deficiency[1][8] and hypopituitarism.[1] Findings affecting pituitary function in some Johanson–Blizzard syndrome patients have included such anomalies as the formation of a glial hamartoma (a neoplasm, or tumor composed of glial cells) on a lobe of the pituitary gland, as well congenital underdevelopment of the anterior pituitary.[13] Growth failure and associated short stature (dwarfism) in Johanson–Blizzard syndrome can be attributed to growth hormone deficiency caused by diminished anterior pituitary function, with malabsorption of fats playing a subsequent role.[1][4][14]
Nasopharyngeal
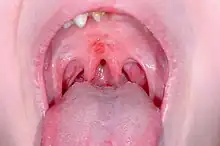
The primary malformation apparent with Johanson–Blizzard syndrome is hypoplasia (underdevelopment) of the nasal alae, or "wing of the nose".[1][2][7] Both hypoplasia and aplasia (partial or complete absence) of structural cartilage and tissue in this area of the nose, along with the underlying alae nasi muscle, are prevailing features of the disorder. Together, these malformations give the nose and nostrils an odd shape and appearance.[7][15]
Neurological
Intellectual disability ranging from mild to severe is present in the majority of Johanson–Blizzard syndrome patients, and is related to the deleterious nature of the known mutagen responsible for the disorder and its effects on the developing central nervous system.[1][6][16] Normal intelligence and age appropriate social development, however, have been reported in a few instances of Johanson–Blizzard syndrome.[12][16]
Auditory
Findings with the inner ear in Johanson–Blizzard syndrome give explanation to the presence of bilateral sensorineural hearing loss in most patients affected by the disorder. The formation of cystic tissue in both the cochlea and vestibule, with resulting dilation (widening) and malformation of these delicate structures has been implicated.[7][9][17] Congenital deformations of the temporal bone and associated adverse anatomical effects on innervation and development of the inner ear also contribute to this type of hearing loss.[17][18]
Craniofacial
Other abnormalities, affecting the scalp, head, face, jaw and teeth may be found with JBS. These include: ectodermal mid-line scalp defects with sparse, oddly-patterned hair growth;[2][9] aplasia cutis (underdeveloped, very thin skin) over the head,[19] an enlarged fontanelle ("soft spot" on the head of young infants),[14] microcephaly (undersized skull),[19] prominent forehead,[14] absence of eyebrows and eyelashes,[14] mongoloid eye shape,[17] nasolacrimo- cutaneous fistulae (this refers to the formation of an abnormal secondary passageway from either the tear duct or lacrimal sac to the facial skin surface, possibly discharging fluid),[9] flattened ears,[14] micrognathism of the maxilla and mandible (underdevelopment of the upper and lower jaw, respectively), with the maxilla more prominently affected in some cases;[14][20][21] congenital clefting of bones surrounding the optical orbit (eye socket), such as the frontal and lacrimal bone;[20] and maldeveloped deciduous teeth ("baby teeth"), with an absence of permanent teeth.[9][14]
Effects on other organ systems
Additional congenital anomalies, effects on other organs, and less common features of JBS have included: imperforate anus (occlusion of the anus),[22] vesicoureteral reflux (reversal of the flow of urine, from the bladder back into the ureters, toward the kidneys);[14] duplex of the uterus and vagina in female infants,[7] neonatal cholestasis of the liver, with cirrhosis and portal hypertension (high blood pressure in the hepatic portal vein);[22] dilated cardiomyopathy,[23] dextrocardia (congenital displacement of the heart to the right side of the chest),[1] atrial and ventricular septal defect;[1] low birth-weight,[24] failure to thrive,[24] hypotonia (decreased muscle tone);[19] sacral hiatus (a structural deficiency of the sacral vertebrae),[24] congenital cataracts,[24] and cafe-au-lait spots.[2]
Genetics
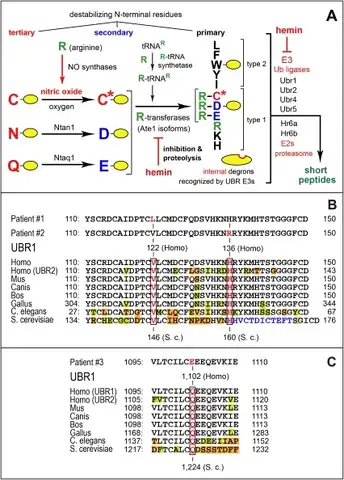 a-c)Arg/N-end rule pathway and missense mutations in human UBR1 that underlie specific cases of Johanson-Blizzard syndrome
a-c)Arg/N-end rule pathway and missense mutations in human UBR1 that underlie specific cases of Johanson-Blizzard syndrome Johanson-Blizzard syndrome has an autosomal recessive pattern of inheritance.
Johanson-Blizzard syndrome has an autosomal recessive pattern of inheritance.
Johanson–Blizzard syndrome has an autosomal recessive pattern of inheritance resulting from loss of function (usually deleterious as nonsense, frameshift, or splice site) mutations in the Ubiquitin-Protein Ligase E3 Component N- Recognin gene (UBR1), which encodes for a specific ubiquitin ligase enzyme.[25] This means the defective UBR1 gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the Johanson–Blizzard syndrome. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Johanson–Blizzard syndrome results from one or multiple mutations in UBR1, specifically at a fixed chromosomal position known as locus 15q15.2 or human chromosome 15, q-arm region 1, band 5, sub-band 2.[26] This gene spans around 161kb (161,000 base pairs) in length and contains 47 exons expressed as mRNA.[27] In comparison, mouse 120kb UBR1 is in the middle of chromosome 2 and shows homology of synteny (co-localized loci in same chromosome) with its human counterpart through its 50 exons. The protein has also been weighed in at 200-kD in mice compared to 225-kD in Saccharomyces cerevisiae.[27][26]
UBR1 encodes one of at least four functionally overlapping E3 ubiquitin ligases of the N-end rule pathway. This pathway consists of a conserved proteolytic system of proteins that destabilize N-terminal residues, meaning UBR1 codes for proteins with degron parts that send degradation signals to the cell, inducing metabolic instability. This specific signal is called N-degron, and its causal set of peptides yields the N-end rule, which relates the protein's in vivo half-life to the identity of its N-terminal residue through a ubiquitin system (N-end rule pathway). The N-recognin, also known as E3, binds to the destabilizing N-terminal residue of a substrate protein to form a substrate-linked miltiubiquitin chain.[26]
The direct connection between UBR1 mutations altering the protein degradation system and specific Johanson–Blizzard syndrome clinical anomalies (symptoms of diagnosis) is still undetermined as origin of possible mutagenic genetic variations varies from just the father alleles to both alleles; and single or multi-exon deletions/duplications in which all 47 UBR1 exons must be taken into account when performing Sanger sequencing and Multiplex ligation-dependent probe amplification (MLPA), meaning there is no obvious candidate gene.[28] However, most certain UBR1 mutations predict premature translational stop codons, with two missense mutations altering residues highly conserved among different species.[27] One of these missense mutations affects a conserved motif important for UBR1 substrate-binding by converting histidine at location 136 to arginine accompanied by intervening sequence. Bidirectional analysis of all 47 exons (including ~20bp flanking intronic regions) reveals homozygous mutation in exon 19, where threonine nucleotide substitutes cysteine, resulting in a missensed serine residue between peptide locations 698 and 702 completely conserved throughout vertebrate UBR1 (even UBR2) protein.[28] Another cysteine to threonine mutation, but homozygous nonsense in nature, has also been confirmed in Johanson–Blizzard syndrome patients with no functional UBR1 protein, but mild symptoms are also common in missense mutations in at least one of the two UBR1 copies with possible residual activity of gene product.[25] 2 Heterozygous mutations from nonconsanguineous parents arise from adenine to guanine conversion at nucleotide 407 resulting in a histidine 136 substitution to arginine at splice donor site.[27] Next non-consanguineous homozygous nonsense mutation happens at glutamine 513 becoming a stop codon by a cytosine to thymine conversion caused by a cytosine to thymine transition at nucleotide 1537 in exon 13.[27] Continuing homozygous mutations, one converts guanine to adenine, in intron 26 resulting in residual normal protein production.[29] The last homozygous mutation turns guanine to adenine in intron 12 by skipping exon 13 through a frameshift and causing premature termination.[30] Maternally inherited heterozygous nonsense mutation of cysteine to adenine resulting in a tyrosine has also been classified at residue 1508.[31] Another heterozygous missense mutation on leucine linked to an arganine at exon 44 is considered pathogenic because leucine at residue 1597 is highly conserved among different species. Lastly, a splice site mutation has been identified intervening sequence changing thymine for cytosine at nucleotide 20.[27]
Pathophysiology
Johanson–Blizzard syndrome is caused by mutations in the UBR1 gene, which encodes one of several ubiquitin ligase enzymes of the N-end rule pathway.[1][6]
The protein ubiquitin is a universal, "ubiquitously" expressed protein common to eukaryotic organisms. Ubiquitin plays a role in the regulation of other proteins by tagging them for eventual degradation by proteasomes.[32] This process begins when ubiquitin ligase covalently attaches a ubiquitin molecule to the lysine side chain of the target protein substrate (the misfolded, damaged, malfunctioning or unneeded protein that needs to be degraded). This is repeated a number of times in succession forming a chain of ubiquitin molecules, which is a process referred to as polyubiquitination. The polyubiquitination of the target protein signals the proteasome to break it down, which it does via proteolysis.[32] The ubiquitin-proteasome system plays a crucial role in the non-lysosomal degradation of intracellular proteins, and ubiquitin can also participate in modifying proteins to perform certain tasks.[32][33][34] Both degradation and modification of proteins within the cell are part of a broader regulatory scheme, necessary for cellular processes such as cell division, cell signaling, cell surface receptor function, apoptosis, DNA maintenance, inflammatory response and developmental quality control associated with the cell cycle and homeostasis in general.[33][34]
Ubiquitin-mediated degradation of proteins occurs through the N-end rule pathway.[35][36] In eukaryotes, including humans, the N-end rule pathway is part of the ubiquitin system.[35] Composed of a highly selective single-residue code (a single amino acid nucleotide sequence), the N-end rule serves as a mechanism which can relate the stability of a protein to the identity of the amino acid at its N-terminus (the end of the polypeptide with an amino group, which in the ubiquitin system may be involved in the reactive destabilization of the protein).[35][36][37]
In JBS, mutations in the UBR1 gene alter, disrupt or prevent the synthesis of ubiquitin ligase.[1][6] In the pancreatic acinar cells, UBR1 is more highly expressed than anywhere else in the body.[1] Impairment of the ubiquitin-proteasome system directly related to insufficient activity of ubiquitin ligase has been established as the cause of both congenital and progressive inflammatory damage, fatty tissue replacement, connective tissue proliferation and errors in innervation of the acini and islets, correlating to failures of normal apoptotic destruction of damaged cells and constitutive malpresence of proteins.[1][3][6] This also applies to other areas affected by deleterious UBR1 expression, such as the craniofacial area, musculoskeletal and nervous systems, dentition and organs.[1][6][22]
Missense, nonsense and splice site mutations of the UBR1 gene in both parents have been found with JBS, confirming the homozygous nature of the JBS phenotype. Variability of the phenotype, associated with residual ubiquitin ligase activity in some patients, has also been attributed to hypomorphic mutations occasionally found in either of the carrier parents.[1][3][6][22][23] The UBR1 gene is located on human chromosome 15.[6]
Diagnosis
Johanson-Blizzard Syndrome may be diagnosed based on the identification of characteristic symptoms or by testing for mutations on the UBR1 gene which are known to case this disorder.[38]
Treatment
While there is no cure for Johanson–Blizzard syndrome, treatment and management of specific symptoms and features of the disorder are applied and can often be successful. Variability in the severity of Johanson–Blizzard syndrome on a case-by-case basis determines the requirements and effectiveness of any treatment selected.
Pancreatic insufficiency and malabsorption can be managed with pancreatic enzyme replacement therapy, such as pancrelipase supplementation and other related methods.[1]
Craniofacial and skeletal deformities may require surgical correction, using techniques including bone grafts and osteotomy procedures.[20] Sensorineural hearing loss can be managed with the use of hearing aids and educational services designated for the hearing impaired.[12][17]
Special education, specialized counseling methods and occupational therapy designed for those with intellectual disabilities have proven to be effective, for both the patient and their families.[39]
Research
Mice that are viable, fertile and lacked substantial phenotypic abnormalities other than reduced weight, with disproportionate decreases in skeletal muscle and adipose tissue are used for their pancreatic sensitive to scretagogue cholecytokinin by knocking out UBR1.This links signaling circuitry between pancreatic enzyme secretion and its source compound controlled by N-end rule pathway, ultimately determining pancreatic homeostasis is influenced by UBR1.[40][27] Saccharomyces cerevisiae also contains regions essential for recognition of the N-end rule substrates by UBR1 protein, as well as rabbits for through reticulocyte tryptic peptides after purification to E3α.[41]
Eponym
Johanson–Blizzard syndrome was named after Ann J. Johanson and Robert M. Blizzard, the pediatricians who first described the disorder in a 1971 journal report.[15][42]
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Alkhouri N, Kaplan B, Kay M, Shealy A, Crowe C, Bauhuber S, Zenker M (Nov 2008). "Johanson-Blizzard syndrome with mild phenotypic features confirmed by UBR1 gene testing". World Journal of Gastroenterology. 14 (44): 6863–6866. doi:10.3748/wjg.14.6863. PMC 2773884. PMID 19058315. Archived from the original (Free full text) on 2012-02-18.
- 1 2 3 4 5 Kulkarni ML, Shetty SK, Kallambella KS, Kulkarni PM (Dec 2004). "Johanson--blizzard syndrome". Indian Journal of Pediatrics. 71 (12): 1127–1129. doi:10.1007/BF02829829. PMID 15630323. S2CID 38967896.
- 1 2 3 4 5 Zenker M, Mayerle J, Reis A, Lerch MM (Jun 2006). "Genetic basis and pancreatic biology of Johanson-Blizzard syndrome". Endocrinology and Metabolism Clinics of North America. 35 (2): 243–253, vii–viii. doi:10.1016/j.ecl.2006.02.013. PMID 16632090.
- 1 2 Sandhu BK, Brueton MJ (November 1989). "Concurrent pancreatic and growth hormone insufficiency in Johanson-Blizzard syndrome". J. Pediatr. Gastroenterol. Nutr. 9 (4): 535–8. doi:10.1097/00005176-198911000-00026. PMID 2621533.
- 1 2 3 Steinbach WJ, Hintz RL (Nov 2000). "Diabetes mellitus and profound insulin resistance in Johanson-Blizzard syndrome". Journal of Pediatric Endocrinology & Metabolism. 13 (9): 1633–1636. doi:10.1515/jpem.2000.13.9.1633. ISSN 0334-018X. PMID 11154160. S2CID 20598132.
- 1 2 3 4 5 6 7 8 Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, Beier M, Hülskamp G, Guzman C, Rehder H, Beemer FA, Hamel B, Vanlieferinghen P, Gershoni-Baruch R, Vieira MW, Dumic M, Auslender R, Gil-Da-Silva-Lopes VL, Steinlicht S, Rauh M, Shalev SA, Thiel C, Ekici AB, Winterpacht A, Kwon YT, Varshavsky A, Reis A (Dec 2005). "Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome)" (PDF). Nature Genetics. 37 (12): 1345–1350. doi:10.1038/ng1681. PMID 16311597. S2CID 23050042. Archived (PDF) from the original on 2022-10-17. Retrieved 2022-05-06.
- 1 2 3 4 5 Rosanowski F, Hoppe U, Hies T, Eysholdt U (Oct 1998). "Johanson-Blizzard syndrome. A complex dysplasia syndrome with aplasia of the nasal alae and inner ear deafness". HNO. 46 (10): 876–878. doi:10.1007/s001060050328. PMID 9846268. S2CID 43526278.
- 1 2 3 4 Takahashi T, Fujishima M, Tsuchida S, Enoki M, Takada G (Aug 2004). "Johanson-blizzard syndrome: loss of glucagon secretion response to insulin-induced hypoglycemia". Journal of Pediatric Endocrinology & Metabolism. 17 (8): 1141–1144. doi:10.1515/jpem.2004.17.8.1141. ISSN 0334-018X. PMID 15379429. S2CID 5658865.
- 1 2 3 4 5 6 Daentl DL, Frías JL, Gilbert EF, Opitz JM (1979). "The Johanson-Blizzard syndrome: case report and autopsy findings". American Journal of Medical Genetics. 3 (2): 129–135. doi:10.1002/ajmg.1320030203. PMID 474625.
- ↑ Jones NL, Hofley PM, Durie PR (Sep 1994). "Pathophysiology of the pancreatic defect in Johanson-Blizzard syndrome: a disorder of acinar development". The Journal of Pediatrics. 125 (3): 406–408. doi:10.1016/S0022-3476(05)83286-X. PMID 8071749.
- 1 2 Nagashima K, Yagi H, Kuroume T (Feb 1993). "A case of Johanson-Blizzard syndrome complicated by diabetes mellitus". Clinical Genetics. 43 (2): 98–100. doi:10.1111/j.1399-0004.1993.tb04458.x. ISSN 0009-9163. PMID 8448911. S2CID 33408299.
- 1 2 3 Gould NS, Paton JB, Bennett AR (Jun 1989). "Johanson-Blizzard syndrome: clinical and pathological findings in 2 sibs". American Journal of Medical Genetics. 33 (2): 194–199. doi:10.1002/ajmg.1320330212. PMID 2669481.
- ↑ Hoffman WH, Lee JR, Kovacs K, Chen H, Yaghmai F (Jan 2007). "Johanson-Blizzard syndrome: autopsy findings with special emphasis on hypopituitarism and review of the literature". Pediatric and Developmental Pathology. 10 (1): 55–60. doi:10.2350/06-05-0085.1. PMID 17378628. S2CID 42630522.
- 1 2 3 4 5 6 7 8 Fichter CR, Johnson GA, Braddock SR, Tobias JD (January 2003). "Perioperative care of the child with the Johanson-Blizzard syndrome". Pediatric Anesthesia. 13 (1): 72–5. doi:10.1046/j.1460-9592.2003.00957.x. PMID 12535044. S2CID 23268410.
- 1 2 Online Mendelian Inheritance in Man (OMIM): 243800
- 1 2 Moeschler JB, Polak MJ, Jenkins JJ, Amato RS (January 1987). "The Johanson-Blizzard syndrome: a second report of full autopsy findings". Am. J. Med. Genet. 26 (1): 133–8. doi:10.1002/ajmg.1320260120. PMID 3812553.
- 1 2 3 4 Braun J, Lerner A, Gershoni-Baruch R (1991). "The temporal bone in the Johanson-Blizzard syndrome. A CT study". Pediatric Radiology. 21 (8): 580–3. doi:10.1007/BF02012603. PMID 1815181. S2CID 27095180.
- ↑ Bamiou DE, Phelps P, Sirimanna T (March 2000). "Temporal bone computed tomography findings in bilateral sensorineural hearing loss". Arch. Dis. Child. 82 (3): 257–60. doi:10.1136/adc.82.3.257. PMC 1718255. PMID 10685935.
- 1 2 3 Mardin MK, Ghandour M, Sakati NA, Nyhan WL (Nov 1978). "Johanson-Blizzard syndrome in a large inbred kindred with three involved members". Clin Genet. 14 (5): 247–250. doi:10.1111/j.1399-0004.1978.tb02141.x. PMID 709902. S2CID 35031493.
- 1 2 3 Kobayashi S, Ohmori K, Sekiguchi J (Sep 1995). "Johanson-Blizzard syndrome facial anomaly and its correction using a microsurgical bone graft and tripartite osteotomy". J Craniofac Surg. 6 (5): 382–385. doi:10.1097/00001665-199509000-00011. PMID 9020718.
- ↑ Motohashi N, Pruzansky S, Day D (1981). "Roentgencephalometric analysis of craniofacial growth in the Johanson-Blizzard syndrome". J Craniofac Genet Dev Biol. 1 (1): 57–72. PMID 7341643.
- 1 2 3 4 Al-Dosari MS, Al-Muhsen S, Al-Jazaeri A, Mayerle J, Zenker M, Alkuraya FS (July 2008). "Johanson-Blizzard syndrome: report of a novel mutation and severe liver involvement". Am J Med Genet A. 146A (14): 1875–9. doi:10.1002/ajmg.a.32401. PMID 18553553. S2CID 30927282.
- 1 2 Elting M, Kariminejad A, de Sonnaville ML, Ottenkamp J, Bauhuber S, Bozorgmehr B, Zenker M, Cobben JM (December 2008). "Johanson-Blizzard syndrome caused by identical UBR1 mutations in two unrelated girls, one with a cardiomyopathy". Am J Med Genet A. 146A (23): 3058–61. doi:10.1002/ajmg.a.32566. PMID 19006206. S2CID 20782358.
- 1 2 3 4 Dumić M, Ille J, Bobonj G, Kordić R, Batinica S (May 1998). "Johanson-Blizzardov sindrom" [The Johanson-Blizzard syndrome]. Lijec Vjesn (in hrvatski). 120 (5): 114–6. PMID 9748788.
- 1 2 Quaio, C. R.; Koda, Y. K.; Bertola, D. R.; Sukalo, M.; Zenker, M.; Kim, C. A. (2014-06-09). "Case report. Johanson-Blizzard syndrome: a report of gender-discordant twins with a novel UBR1 mutation". Genetics and Molecular Research. 13 (2): 4159–4164. doi:10.4238/2014.June.9.2. ISSN 1676-5680. PMID 25036160.
- 1 2 3 Kwon, Y. T.; Reiss, Y.; Fried, V. A.; Hershko, A.; Yoon, J. K.; Gonda, D. K.; Sangan, P.; Copeland, N. G.; Jenkins, N. A.; Varshavsky, A. (1998-07-07). "The mouse and human genes encoding the recognition component of the N-end rule pathway". Proceedings of the National Academy of Sciences of the United States of America. 95 (14): 7898–7903. Bibcode:1998PNAS...95.7898K. doi:10.1073/pnas.95.14.7898. ISSN 0027-8424. PMC 20901. PMID 9653112.
- 1 2 3 4 5 6 7 Zenker, Martin; Mayerle, Julia; Lerch, Markus M.; Tagariello, Andreas; Zerres, Klaus; Durie, Peter R.; Beier, Matthias; Hülskamp, Georg; Guzman, Celina; Rehder, Helga; Beemer, Frits A. (December 2005). "Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome)" (PDF). Nature Genetics. 37 (12): 1345–1350. doi:10.1038/ng1681. ISSN 1061-4036. PMID 16311597. S2CID 23050042. Archived (PDF) from the original on 2022-10-17. Retrieved 2022-05-06.
- 1 2 Almashraki, Nabeel; Abdulnabee, Mukarram Zainuddin; Sukalo, Maja; Alrajoudi, Abdullah; Sharafadeen, Iman; Zenker, Martin (2011-10-07). "Johanson-Blizzard syndrome". World Journal of Gastroenterology. 17 (37): 4247–4250. doi:10.3748/wjg.v17.i37.4247. ISSN 2219-2840. PMC 3208372. PMID 22072859.
- ↑ Elting, Mariet; Kariminejad, Ariana; de Sonnaville, Marie-Louise; Ottenkamp, Jaap; Bauhuber, Susanne; Bozorgmehr, Bita; Zenker, Martin; Cobben, Jan M. (2008-12-01). "Johanson-Blizzard syndrome caused by identical UBR1 mutations in two unrelated girls, one with a cardiomyopathy". American Journal of Medical Genetics. Part A. 146A (23): 3058–3061. doi:10.1002/ajmg.a.32566. ISSN 1552-4833. PMID 19006206. S2CID 20782358.
- ↑ Al-Dosari, Mohammed S.; Al-Muhsen, Saleh; Al-Jazaeri, Ayman; Mayerle, Julia; Zenker, Martin; Alkuraya, Fowzan S. (2008-07-15). "Johanson-Blizzard syndrome: report of a novel mutation and severe liver involvement". American Journal of Medical Genetics. Part A. 146A (14): 1875–1879. doi:10.1002/ajmg.a.32401. ISSN 1552-4833. PMID 18553553. S2CID 30927282.
- ↑ Sukalo, Maja; Fiedler, Ariane; Guzmán, Celina; Spranger, Stephanie; Addor, Marie-Claude; Mcheik, Jiad N.; Oltra Benavent, Manuel; Cobben, Jan M.; Gillis, Lynette A.; Shealy, Amy G.; Deshpande, Charu (May 2014). "Mutations in the Human UBR1 Gene and the Associated Phenotypic Spectrum". Human Mutation. 35 (5): 521–531. doi:10.1002/humu.22538. PMID 24599544. S2CID 25288051.
- 1 2 3 Wang J, Maldonado MA (August 2006). "The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases". Cell Mol Immunol. 3 (4): 255–61. PMID 16978533.
- 1 2 Ciechanover A (September 1994). "The ubiquitin-mediated proteolytic pathway: mechanisms of action and cellular physiology". Biol Chem Hoppe-Seyler. 375 (9): 565–81. doi:10.1515/bchm3.1994.375.8.565. PMID 7840898.
- 1 2 Ciechanover A, Iwai K (April 2004). "The ubiquitin system: from basic mechanisms to the patient bed". IUBMB Life. 56 (4): 193–201. doi:10.1080/1521654042000223616. PMID 15230346. S2CID 25409332.
- 1 2 3 Varshavsky A (January 1997). "The N-end rule pathway of protein degradation". Genes Cells. 2 (1): 13–28. doi:10.1046/j.1365-2443.1997.1020301.x. PMID 9112437. S2CID 27736735.
- 1 2 Baker RT, Varshavsky A (February 1991). "Inhibition of the N-end rule pathway in living cells". Proc Natl Acad Sci USA. 88 (4): 1090–4. Bibcode:1991PNAS...88.1090B. doi:10.1073/pnas.88.4.1090. PMC 50962. PMID 1899923.
- ↑ Gonda DK, Bachmair A, Wünning I, Tobias JW, Lane WS, Varshavsky A (October 1989). "Universality and structure of the N-end rule". J Biol Chem. 264 (28): 16700–12. doi:10.1016/S0021-9258(19)84762-2. PMID 2506181.
- ↑ "Johanson-Blizzard Syndrome". National Organisation for Rare Disorders. Archived from the original on June 13, 2021. Retrieved June 13, 2021.
- ↑ Prater JF, D'Addio K (March 2002). "Johanson-Blizzard syndrome--a case study, behavioral manifestations, and successful treatment strategies". Biol Psychiatry. 51 (6): 515–7. doi:10.1016/S0006-3223(01)01337-3. PMID 11922888. S2CID 10377190.
- ↑ Kwon, Y. T.; Xia, Z.; Davydov, I. V.; Lecker, S. H.; Varshavsky, A. (December 2001). "Construction and analysis of mouse strains lacking the ubiquitin ligase UBR1 (E3alpha) of the N-end rule pathway". Molecular and Cellular Biology. 21 (23): 8007–8021. doi:10.1128/MCB.21.23.8007-8021.2001. ISSN 0270-7306. PMC 99968. PMID 11689692.
- ↑ Kwon, Y. T.; Reiss, Y.; Fried, V. A.; Hershko, A.; Yoon, J. K.; Gonda, D. K.; Sangan, P.; Copeland, N. G.; Jenkins, N. A.; Varshavsky, A. (1998-07-07). "The mouse and human genes encoding the recognition component of the N-end rule pathway". Proceedings of the National Academy of Sciences. 95 (14): 7898–7903. Bibcode:1998PNAS...95.7898K. doi:10.1073/pnas.95.14.7898. ISSN 0027-8424. PMC 20901. PMID 9653112.
- ↑ Johanson A, Blizzard R (December 1971). "A syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsorption". J Pediatr. 79 (6): 982–7. doi:10.1016/S0022-3476(71)80194-4. PMID 5171616.
External links
| Classification | |
|---|---|
| External resources |
|