B-cell prolymphocytic leukemia
| B-cell prolymphocytic leukemia | |
|---|---|
 | |
| Prolymphocyte | |
| Specialty | Hematology, oncology |
B-cell prolymphocytic leukemia, referred to as B-PLL, is a rare blood cancer. It is a more aggressive, but still treatable, form of leukemia.
Specifically, B-PLL is a prolymphocytic leukemia (PLL) that affects prolymphocytes – immature forms of B-lymphocytes and T-lymphocytes – in the peripheral blood, bone marrow, and spleen. It is an aggressive cancer that presents poor response to treatment.[1]Mature lymphocytes are infection-fighting immune system cells. B-lymphocytes have two responsibilities:[2]
- Production of antibodies – In response to antigens, B-lymphocytes produce and release antibodies specific to foreign substances in order to aid in their identification and elimination phagocytes
- Generation of memory cells – Interactions between antibodies and antigens allow B-lymphocytes to establish cellular memories, otherwise known as immunities that allow the body to respond more rapidly and efficiently to previously encountered species
Classification
It is categorized as a lymphoproliferative disorder due to the excessive production of lymphocytes, in B-PLL there is excess production of B-prolymphocytes by the bone marrow. These immature lymphocytes are not normally found in the blood; part of their maturation process is being programmed to produce antibodies against foreign material prior to their departure from the bone marrow.[3] In B-PLL, malignant B-prolymphocytes disrupt the adaptive capabilities of the immune system due to the lack of mature B-lymphocytes.
It has been suggested that some cases may represent a variant of mantle cell lymphoma.[4]
Signs and symptoms
This type of leukemia is characterized by:[5][6]
- More than 55% of circulating cells in peripheral blood (red blood cells, white blood cells and platelets collectively) are prolymphocytes. Generally, prolymphocyte proportion exceeds 90%
- Minimal or absence of lymphadenopathy – abnormalities in size, number or consistency of lymph nodes
- Splenomegaly - Abnormal enlargement of the spleen
- High white blood cell count
- B-symptoms – Fever, night sweats and/or weight loss
Similar to other leukemias, B-cell prolymphocytic leukemia is often asymptomatic. The most common signs and symptoms are the result of the inability of the bone marrow to produce normal levels of blood cells:[7]
Diagnosis
Diagnosis of B-PLL is difficult due to its considerable overlap with other mature B-cell leukemias and lymphomas.[8] It requires integration of morphology with diagnostic tests including immunophenotyping and chromosome analysis (cytogenetics).
Morphology
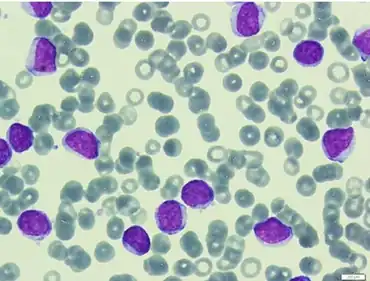
The malignant B cells are larger than average.
In order to diagnose a patient with B-PLL, b-prolymphocyte composition of a patient's blood cells must exceed 55%. High white blood cell counts – greater than 100 x 109/L [8] – are also indicative of B-PLL. B-prolymphocytes are characterized by:[8][9][10]
- Large size – approximately twice the size of a normal small lymphocyte
- Round or oval-shaped nuclei
- Single prominent nucleolus
- Moderately condensed nuclear chromatin
- High nuclear-cytoplasmic ratio – indicates more abundant cytoplasm
Immunophenotype
This technique is used to study proteins expressed in cells using immunologic markers. In B-PLL patients there is strong expression of surface immunoglobulin – a membrane-bound form of an antibody, b-lymphocyte surface antigens CD19, CD20, CD22, CD79a and FMC7, and weak expression of CD5 and CD23.[11] Due to the similarities among lymphoproliferative disorders, it is often difficult to diagnose patients. Immunophenotyping helps distinguish B-PLL from similar diseases, one of its key identifiers is the absence in expression of the surface antigens CD10, CD11c, CD25, CD103 and cyclin D1 – an important regulator of cell-cycle progression.[5]
A case has been described as CD20+, CD22+, and CD5-.[12]
It can also be CD5+.[13]
Another case was described as CD45+, CD19+, CD20+, CD5+, HLA-DR+, CD10-, CD23+/-, CD38+ and FMC7-.[14]
Cytogenetics
B-PLL is rare, consequently few genetic studies have focused on this disease. As a result, the associated genetic lesions underlying B-PLL are largely unknown.
Chromosomal Mutations
The most commonly reported abnormalities have occurred at chromosome 14, specifically in a region of the chromosome called band q23 (14q23). Translocations to this location lead to overexpression of the cyclin D1 gene[11] which has been linked to both the development and progression of a number of cancers.[15] Other chromosomal abnormalities have been reported on 6q21, 11q23, 12p12, 13q14 and 17p.[11]
It can involve deletions from chromosome 11 and chromosome 13.[16]
TP53 Gene
Among the documented studies, mutations to the TP53 gene have occurred in 75% of all cases of B-PLL. This is the highest incidence among all sub-types of B-cell malignancies. Mutations to this gene have also been documented in other hematologic malignancies.[17]
TP53 is an important transcriptional activator of genes involved in the regulation of the G1 checkpoint of the cell cycle as well as certain genes responsible for programmed-cell death (apoptosis). It is believed that mutations to TP53 are responsible for the frequent therapy resistance and aggressive course of this disease.[17]
c-MYC Gene
In a small number of B-PLL cases, abnormalities in the c-MYC gene have been observed. It is considered a global amplifier and influences nearly all aspects of cellular activity. Among the number of genes it regulates, most are involved in cell growth, cell cycle progression, protein biosynthesis and apoptosis. Amplification of c-MYC has been reported in B-PLL patients and while the consequences are unclear, it is generally associated with poor clinical outcome.[18]
Biopsy
After physicians have identified an abnormality in the composition of the peripheral blood, biopsies (tissue samples) from a patient's bone marrow and/or spleen are often recommended for confirmation. A bone marrow biopsy involves the removal of a small amount of tissue that is further analyzed for abnormalities,[19] for B-PLL pathologists look for prolymphocytic infiltration where the hematopoietic stem cells of the bone marrow are replaced with prolymphocytes due to excess production. In 50% of reported cases, it was common for patients to be both anemic (lack healthy red blood cells in blood) and thrombocytopenic (deficiency of platelets in blood).[20]
Treatment
The rarity of B-PLL paired with its considerably fast progression compared to other leukemias has resulted in difficult production of effective treatments. This disease is currently incurable, treatments and therapy are guided to reduce prolymphocyte abundance in the blood and production by the bone marrow, treating symptoms and controlling progression.[20]
Watchful Waiting
Some patients do not require immediate treatment after diagnosis; these patients include those that do not show overt symptoms or whose cancer has not been observed to be progressing. Regular check-ups with physicians are required to actively monitor the patient's condition; once there is evidence of disease progression or patient distress from symptoms, treatment will be implemented.[7][21]
Chemotherapy
B-PLL has a very aggressive clinical course and refractoriness to chemotherapy;[8] it is believed this resistance is the result of mutations to the TP53 gene. Its resistant nature has led to the use of combinations of chemotherapy drugs. Drug regimens recommended and employed by physicians are unique to each patient and are based on previous chemotherapy experience along with potential side effects. In addition to the utilization of combinations of chemotherapeutic drugs, it is most often paired with immunotherapy treatments.[8]
Targeted Therapy
Monoclonal Antibodies
A type of targeted therapy that recognizes specific proteins in leukemia cells preventing collateral damage to normal, healthy cells.[19] The following are compounds currently showing promising results in clinical trials and studies:
- Rituximab is a widely used monoclonal antibody in treating B-cell malignancies, it is directed against the surface protein CD20. Case studies have documented successful treatment of B-PLL solely with rituximab; additional studies have reported positive activity when rituximab is paired with the chemotherapeutic drugs fludarabine or bendamustine together with the anthracyclines mitoxantrone or epirubicin[10]
- Alemtuzumab is a humanized antibody that targets the CD52 antigen which is highly expressed in malignant B-lymphocytes. In vitro tests have demonstrated that it induces cell death. Furthermore, it is most active in the blood, bone marrow and spleen, all of which are main sites involved with B-PLL and thus could serve as a potential agent in treating this disease with more research[10]
Splenectomy or Radiation Therapy to Spleen
Patients with splenomegaly (enlarged spleen), unfit for systemic treatment or refractive to chemotherapy may have their spleens removed via splenectomy or undergo splenic irradiation in order to relieve pain, control their symptoms, and allow removal of a major proliferative focus and tumour bulk in this disease.[8][21]
Splenic irradiation has been used in the treatment.[22]
Stem Cell Transplantation
A stem cell transplant is a procedure that uses highly specialized cells called hematopoietic stem cells to replace bone marrow that contains the leukemia. This procedure should be considered in younger patients that have responded well to initial treatments because the progression and spread of this disease is inevitable.[8] However, stem cell transplantation is a high-risk procedure, with significant morbidity and mortality rates. Furthermore, it is often not a feasible option due to the presence of other systemic diseases/conditions.[8][19]
Prognosis
Despite advancements in treatments and deeper understanding of pathogenesis, the prognosis for B-PLL patients is poor[23] , with early relapse and median survival time between 3–5 years.[24][25]
Epidemiology
B-PLL represents less than 1% of all leukemia cases worldwide,[26] mainly affecting the elderly population with a mean age of presentation between 65 and 70[27] years. Most cases have shown slight male predominance, with a male-to-female ratio of 1.6 to 1,[26] and the vast majority of patients being Caucasians.[5]
References
- ↑ "France - Lymphoproliferative Syndrome B-Cell Prolymphocytic Leukemia |". icgc.org. Archived from the original on 2016-10-27. Retrieved 2016-11-18.
- ↑ "The Immune System". www.nobelprize.org. Archived from the original on 2016-11-07. Retrieved 2016-11-20.
- ↑ "The Immune System and Primary Immunodeficiency | Immune Deficiency Foundation". primaryimmune.org. Archived from the original on 2022-01-26. Retrieved 2016-11-20.
- ↑ Ruchlemer R, Parry-Jones N, Brito-Babapulle V, et al. (May 2004). "B-prolymphocytic leukaemia with t(11;14) revisited: a splenomegalic form of mantle cell lymphoma evolving with leukaemia". Br. J. Haematol. 125 (3): 330–6. doi:10.1111/j.1365-2141.2004.04913.x. PMID 15086413.
- 1 2 3 "B cell prolymphocytic leukemia". www.uptodate.com. Archived from the original on 2017-02-26. Retrieved 2016-10-30.
- ↑ Velden, Vincent H. J. van der; Hoogeveen, Patricia G.; Ridder, Dick de; Struijk, Magdalena Schindler-van der; Zelm, Menno C. van; Sanders, Mathijs; Karsch, Dennis; Beverloo, H. Berna; Lam, King (2014-07-17). "B-cell prolymphocytic leukemia: a specific subgroup of mantle cell lymphoma". Blood. 124 (3): 412–419. doi:10.1182/blood-2013-10-533869. ISSN 0006-4971. PMID 24891323.
- 1 2 "Prolymphocytic leukaemia | Leukaemia CARE". www.leukaemiacare.org.uk. Archived from the original on 2016-12-27. Retrieved 2016-10-30.
- 1 2 3 4 5 6 7 8 Dearden, Claire (2012-07-19). "How I treat prolymphocytic leukemia". Blood. 120 (3): 538–551. doi:10.1182/blood-2012-01-380139. ISSN 0006-4971. PMID 22649104. S2CID 5312650.
- ↑ Wiernik, Peter; Goldman, John; Dutcher, Janice; Kyle, Robert (2012). Neoplastic Diseases of the Blood. Springer. p. 84. doi:10.1007/978-1-4614-3764-2. ISBN 9781461437635.
- 1 2 3 Dearden, Claire (2012-12-08). "B- and T-cell prolymphocytic leukemia: antibody approaches". ASH Education Program Book. 2012 (1): 645–651. doi:10.1182/asheducation.V2012.1.645.3798657. ISSN 1520-4391. PMID 23233647. Archived from the original on 2019-05-17. Retrieved 2021-11-21.
- 1 2 3 Ravandi, Farhad; O'Brien, Susan (December 2005). "Chronic Lymphoid Leukemias Other Than Chronic Lymphocytic Leukemia: Diagnosis and Treatment". Mayo Clinic Proceedings. 80 (12): 1660–1674. doi:10.4065/80.12.1660. PMID 16342661.
- ↑ Yamamoto K, Hamaguchi H, Nagata K, Shibuya H, Takeuchi H (April 1998). "Splenic irradiation for prolymphocytic leukemia: is it preferable as an initial treatment or not?". Jpn. J. Clin. Oncol. 28 (4): 267–9. doi:10.1093/jjco/28.4.267. PMID 9657013.
- ↑ "Pathology". Archived from the original on 7 February 2009. Retrieved 2009-01-31.
- ↑ Crisostomo RH, Fernandez JA, Caceres W (May 2007). "Complex karyotype including chromosomal translocation (8;14) (q24;q32) in one case with B-cell prolymphocytic leukemia". Leuk. Res. 31 (5): 699–701. doi:10.1016/j.leukres.2006.06.010. PMID 16997373.
- ↑ Alao, John P. (2007-01-01). "The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention". Molecular Cancer. 6: 24. doi:10.1186/1476-4598-6-24. ISSN 1476-4598. PMC 1851974. PMID 17407548.
- ↑ Lens D, Matutes E, Catovsky D, Coignet LJ (2000). "Frequent deletions at 11q 23 and 13q14 in B cell prolymphocytic leukemia (B-PLL)". Leukemia. 14 (3): 427–30. doi:10.1038/sj.leu.2401644. PMID 10720137.
- 1 2 Lens, Daniela; Schouwer, Pierre J. J. C. De; Hamoudi, Rifat A.; Abdul-Rauf, Munah; Farahat, Nahla; Matutes, Estella; Crook, Tim; Dyer, Martin J. S.; Catovsky, Daniel (1997-03-15). "p53 Abnormalities in B-Cell Prolymphocytic Leukemia". Blood. 89 (6): 2015–2023. doi:10.1182/blood.V89.6.2015. ISSN 0006-4971. PMID 9058723. Archived from the original on 2022-01-26. Retrieved 2021-11-21.
- ↑ Flatley, Ellen; Chen, Andy I.; Zhao, Xiangrong; Jaffe, Elaine S.; Dunlap, Jennifer B.; Pittaluga, Stefania; Abdullah, Shahed; Olson, Susan B.; Spurgeon, Stephen E. (2014-09-01). "Aberrations of MYC Are a Common Event in B-Cell Prolymphocytic Leukemia". American Journal of Clinical Pathology. 142 (3): 347–354. doi:10.1309/AJCPUBHM8U7ZFLOB. ISSN 0002-9173. PMID 25125625.
- 1 2 3 "Leukemia - B-cell Prolymphocytic Leukemia and Hairy Cell Leukemia: Treatment Options | Cancer.Net". Cancer.Net. 2012-06-25. Archived from the original on 2016-08-13. Retrieved 2016-10-30.
- 1 2 "Zosyn: B-prolymphocytic leukemia: A case study at The Medical Dictionary". the-medical-dictionary.com. Archived from the original on 2019-05-17. Retrieved 2016-11-19.
- 1 2 "Prolymphocytic leukemias (PLLs) - Canadian Cancer Society". www.cancer.ca. Archived from the original on 2017-04-30. Retrieved 2016-10-30.
- ↑ Nakashima H, Saito B, Ariizumi H, Matsuda I, Nakamaki T, Tomoyasu S (December 2008). "Splenic irradiation as a successful treatment for an elderly patient with B-cell prolymphocytic leukemia". Rinsho Ketsueki. 49 (12): 1619–22. doi:10.11406/rinketsu.49.1619. PMID 19110524.
- ↑ Del Giudice I, Davis Z, Matutes E, et al. (2006). "IgVH genes mutation and usage, ZAP-70 and CD38 expression provide new insights on B-cell prolymphocytic leukemia (B-PLL)". Leukemia. 20 (7): 1231–7. doi:10.1038/sj.leu.2404238. PMID 16642047.
- ↑ Armitage, James (2004). Atlas of Clinical Hematology. Philadelphia: Lippincott Williams & Wilkins. p. 84. ISBN 9780781751285.
- ↑ Raghavan, Derek; Blanke, Charles; Johnson, David; Moots, Paul; Reaman, Gregory; Rose, Peter; Sekeres, Mikkael (2012). Textbook of Uncommon Cancer. Journal of the Royal College of Physicians of London. Vol. 22. Wiley-Blackwell. p. 617. doi:10.1002/9781118464557. ISBN 9781118083734. PMC 5379276.
- 1 2 "B-cell Prolymphocytic Leukemia (B-PLL) | Chronic Lymphocytic Leukemia". www.knowcancer.com. Archived from the original on 2016-08-06. Retrieved 2016-10-30.
- ↑ Melo, J. V.; Catovsky, D.; Galton, D. A. (1986-06-01). "The relationship between chronic lymphocytic leukaemia and prolymphocytic leukaemia. I. Clinical and laboratory features of 300 patients and characterization of an intermediate group". British Journal of Haematology. 63 (2): 377–387. doi:10.1111/j.1365-2141.1986.tb05563.x. ISSN 0007-1048. PMID 3487341. Archived from the original on 2018-04-08. Retrieved 2021-11-21.
External links
| Classification |
|---|