Germline mutation
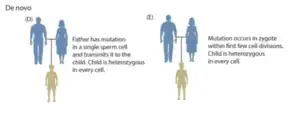
A germline mutation, or germinal mutation, is any detectable variation within germ cells (cells that, when fully developed, become sperm and ova).[1] Mutations in these cells are the only mutations that can be passed on to offspring, when either a mutated sperm or oocyte come together to form a zygote.[2] After this fertilization event occurs, germ cells divide rapidly to produce all of the cells in the body, causing this mutation to be present in every somatic and germline cell in the offspring; this is also known as a constitutional mutation.[2] Germline mutation is distinct from somatic mutation.
Germline mutations can be caused by a variety of endogenous (internal) and exogenous (external) factors, and can occur throughout zygote development.[3] A mutation that arises only in germ cells can result in offspring with a genetic condition that is not present in either parent; this is because the mutation is not present in the rest of the parents' body, only the germline.[3] Due to many severe diseases stemming from de novo germline mutations, different gene editing techniques can be used to induce DNA breaks and repair the mutation.[4]
When mutagenesis occurs
Germline mutations can occur before fertilization and during various stages of zygote development.[3] When the mutation arises will determine the effect it has on offspring. If the mutation arises in either the sperm or the oocyte before development, then the mutation will be present in every cell in the individual's body.[5] A mutation that arises soon after fertilization, but before germline and somatic cells are determined, then the mutation will be present in a large proportion of the individual's cell with no bias towards germline or somatic cells, this is also called a gonosomal mutation.[5] A mutation that arises later in zygote development will be present in a small subset of either somatic or germline cells, but not both.[3][5]
Causes
Endogenous factors
A germline mutation often arises due to endogenous factors, like errors in cellular replication and oxidative damage.[6] This damage is rarely repaired imperfectly, but due to the high rate of germ cell division, can occur frequently.[6]
Endogenous mutations are more prominent in sperm than in ova.[7] This is because spermatocytes go through a larger number of cell divisions throughout a male’s life, resulting in more replication cycles that could result in a DNA mutation.[6] Errors in maternal ovum also occur, but at a lower rate than in paternal sperm.[6] The types of mutations that occur also tend to vary between the sexes.[8] A mother's eggs, after production, remain in stasis until each is utilized in ovulation. This long stasis period has been shown to result in a higher number of chromosomal and large sequence deletions, duplications, insertions, and transversions.[8] The father’s sperm, on the other hand, undergoes continuous replication throughout his lifetime, resulting in many small point mutations that result from errors in replication. These mutations include single base pair deletions, insertions, duplications, and amino acid changes.[7]
Oxidative damage is another endogenous factor that can cause germline mutations. This type of damage is caused by reactive oxygen species that build up in the cell as a by-product of cellular respiration.[9] These reactive oxygen species are missing an electron, and because they are highly electronegative (have a strong electron pull) they will rip an electron away from another molecule.[9] This can initiate DNA damage because it causes the nucleic acid guanine to shift to 8-oxoguanine (8-oxoG). This 8-oxoG molecule is then mistaken for a thymine by DNA polymerase during replication, causing a G>T transversion on one DNA strand, and a C>A transversion on the other.[10]
Exogenous factors
A germline mutation can also occur due to exogenous factors. Similar to somatic mutations, germline mutations can be caused by exposure to harmful substances, which damage the DNA of germ cells. This damage can then either be repaired perfectly, and no mutations will be present, or repaired imperfectly, resulting in a variety of mutations.[11] Exogenous mutagens include harmful chemicals and ionizing radiation; the major difference between germline mutations and somatic mutations is that germ cells are not exposed to UV radiation, and thus not often directly mutated in this manner.[12][13]
Clinical implications
Different germline mutations can affect an individual differently depending on the rest of their genome. A dominant mutation only requires 1 mutated gene to produce the disease phenotype, while a recessive mutation requires both alleles to be mutated to produce the disease phenotype.[14] For example, if the embryo inherits an already mutated allele from the father, and the same allele from the mother underwent an endogenous mutation, then the child will display the disease related to that mutated gene, even though only 1 parent carries the mutant allele.[14] This is only one example of how a child can display a recessive disease while a mutant gene is only carried by one parent.[14] Detection of chromosomal abnormalities can be found in utero for certain diseases by means of blood samples or ultrasound, as well as invasive procedures such as an amniocentesis. Later detection can be found by genome screening.
Cancer
Mutations in tumour suppressor genes or proto-oncogenes can predispose an individual to developing tumours.[15] It is estimated that inherited genetic mutations are involved in 5-10% of cancers. [16] These mutations make a person susceptible to tumour development if the other copy of the oncogene is randomly mutated. These mutations can occur in germ cells, allowing them to be heritable.[15] Individuals who inherit germline mutations in TP53 are predisposed to certain cancer variants because the protein produced by this gene suppresses tumors. Patients with this mutation are also at a risk for Li–Fraumeni syndrome.[16] Other examples include mutations in the BRCA1 and BRCA2 genes which predispose to breast and ovarian cancer, or mutations in MLH1 which predispose to hereditary non-polyposis colorectal cancer.
Huntington's disease
Huntington's disease is an autosomal dominant mutation in the HTT gene. The disorder causes degradation in the brain, resulting in uncontrollable movements and behavior.[17] The mutation involves an expansion of repeats in the Huntington protein, causing it to increase in size. Patients who have more than 40 repeats will most likely be affected. The onset of the disease is determined by the amount of repeats present in the mutation; the greater the number of repeats, the earlier symptoms of the disease will appear.[17][18] Because of the dominant nature of the mutation, only one mutated allele is needed for the disease to be in effect. This means that if one parent is infected, the child will have a 50% chance of inheriting the disease.[19] This disease does not have carriers because if a patient has one mutation, they will (most likely) be affected. The disease typically has a late onset, so many parents have children before they know they have the mutation. The HTT mutation can be detected through genome screening.
Trisomy 21
Trisomy 21 (also known as Down syndrome) results from a child having 3 copies of chromosome 21.[20] This chromosome duplication occurs during germ cell formation, when both copies of chromosome 21 end up in the same daughter cell in either the mother or father, and this mutant germ cell participates in fertilization of the zygote.[20] Another, more common way this can occur is during the first cell division event after the formation of the zygote.[20] The risk of Trisomy 21 increases with maternal age with the risk being 1/2000 (0.05%) at age 20 increasing to 1/100 (1%) at age 40.[21] This disease can be detected by non-invasive as well as invasive procedures prenatally. Non-invasive procedures include scanning for fetal DNA through maternal plasma via a blood sample.[22]
Cystic fibrosis
Cystic fibrosis is an autosomal recessive disorder that causes a variety of symptoms and complications, the most common of which is a thick mucus lining in lung epithelial tissue due to improper salt exchange, but can also affect the pancreas, intestines, liver, and kidneys.[23][24] Many bodily processes can be affected due to the hereditary nature of this disease; if the disease is present in the DNA of both the sperm and the egg, then it will be present in essentially every cell and organ in the body; these mutations can occur initially in the germline cells, or be present in all parental cells.[23] The most common mutation seen in this disease is ΔF508, which means a deletion of the amino acid at the 508 position.[25] If both parents have a mutated CFTR (cystic fibrosis transmembrane conductance regulator) protein, then their children have a 25% of inheriting the disease.[23] If a child has 1 mutated copy of CFTR, they will not develop the disease, but will become a carrier of the disease.[23] The mutation can be detected before birth through amniocentesis, or after birth via prenatal genetic screening. [26]
Current therapies
Many Mendelian disorders stem from dominant point mutations within genes, including cystic fibrosis, beta-thalassemia, sickle-cell anemia, and Tay–Sachs disease.[14] By inducing a double stranded break in sequences surrounding the disease-causing point mutation, a dividing cell can use the non-mutated strand as a template to repair the newly broken DNA strand, getting rid of the disease-causing mutation.[27] Many different genome editing techniques have been used for genome editing, and especially germline mutation editing in germ cells and developing zygotes; however, while these therapies have been extensively studied, their use in human germline editing is limited.[28]
CRISPR/Cas9 editing
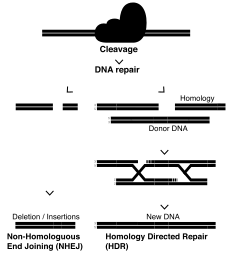
This editing system induces a double stranded break in the DNA, using a guide RNA and effector protein Cas9 to break the DNA backbones at specific target sequences.[27] This system has shown a higher specificity than TALENs or ZFNs due to the Cas9 protein containing homologous (complementary) sequences to the sections of DNA surrounding the site to be cleaved.[27] This broken strand can be repaired in 2 main ways: homologous directed repair (HDR) if a DNA strand is present to be used as a template (either homologous or donor), and if one is not, then the sequence will undergo non-homologous end joining (NHEJ).[27] NHEJ often results in insertions or deletions within the gene of interest, due to the processing of the blunt strand ends, and is a way to study gene knockouts in a lab setting.[4] This method can be used to repair a point mutation by using the sister chromosome as a template, or by providing a double stranded DNA template with the CRISPR/Cas9 machinery to be used as the repair template.[27]
This method has been used in both human and animal models (Drosophila, Mus musculus, and Arabidopsis), and current research is being focused on making this system more specific to minimize off-target cleavage sites.[29]
TALEN editing
The TALEN (transcription activator-like effector nucleases) genome editing system is used to induce a double-stranded DNA break at a specific locus in the genome, which can then be used to mutate or repair the DNA sequence.[30] It functions by using a specific repeated sequence of an amino acid that is 33-34 amino acids in length.[30] The specificity of the DNA binding site is determined by the specific amino acids at positions 12 and 13 (also called the Repeat Variable Diresidue (RVD)) of this tandem repeat, with some RVDs showing a higher specificity for specific amino acids over others.[31] Once the DNA break is initiated, the ends can either be joined with NHEJ that induces mutations, or by HDR that can fix mutations.[27]
ZFN editing
Similar to TALENs, zinc finger nucleases (ZFNs) are used to create a double stranded break in the DNA at a specific locus in the genome.[30] The ZFN editing complex consists of a zinc finger protein (ZFP) and a restriction enzyme cleavage domain.[32] The ZNP domain can be altered to change the DNA sequence that the restriction enzyme cuts, and this cleavage event initiates cellular repair processes, similar to that of CRISPR/Cas9 DNA editing.[32]
Compared to CRISPR/Cas9, the therapeutic applications of this technology are limited, due to the extensive engineering required to make each ZFN specific to the desired sequence.[32]
See also
- Somatic mutation
- Genome Editing
References
- ↑ "NCI Dictionary of Cancer Terms". National Cancer Institute. 2011-02-02. Retrieved 2017-11-30.
- 1 2 Griffiths AJ, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM (2000). "Somatic versus germinal mutation". An Introduction to Genetic Analysis (7th ed.).
- 1 2 3 4 Foulkes WD, Real FX (April 2013). "Many mosaic mutations". Current Oncology. 20 (2): 85–7. doi:10.3747/co.20.1449. PMC 3615857. PMID 23559869.
- 1 2 Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F (January 2014). "Genome-scale CRISPR-Cas9 knockout screening in human cells". Science. 343 (6166): 84–87. Bibcode:2014Sci...343...84S. doi:10.1126/science.1247005. PMC 4089965. PMID 24336571.
- 1 2 3 Samuels ME, Friedman JM (April 2015). "Genetic mosaics and the germ line lineage". Genes. 6 (2): 216–37. doi:10.3390/genes6020216. PMC 4488662. PMID 25898403.
- 1 2 3 4 Crow JF (October 2000). "The origins, patterns and implications of human spontaneous mutation". Nature Reviews Genetics. 1 (1): 40–7. doi:10.1038/35049558. PMID 11262873. S2CID 22279735.
- 1 2 Wong WS, Solomon BD, Bodian DL, Kothiyal P, Eley G, Huddleston KC, Baker R, Thach DC, Iyer RK, Vockley JG, Niederhuber JE (January 2016). "New observations on maternal age effect on germline de novo mutations". Nature Communications. 7: 10486. Bibcode:2016NatCo...710486W. doi:10.1038/ncomms10486. PMC 4735694. PMID 26781218.
- 1 2 Hassold T, Hunt P (December 2009). "Maternal age and chromosomally abnormal pregnancies: what we know and what we wish we knew". Current Opinion in Pediatrics. 21 (6): 703–8. doi:10.1097/MOP.0b013e328332c6ab. PMC 2894811. PMID 19881348.
- 1 2 Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ (September 2003). "Production of reactive oxygen species by mitochondria: central role of complex III". The Journal of Biological Chemistry. 278 (38): 36027–31. doi:10.1074/jbc.M304854200. PMID 12840017.
- ↑ Ohno M, Sakumi K, Fukumura R, Furuichi M, Iwasaki Y, Hokama M, Ikemura T, Tsuzuki T, Gondo Y, Nakabeppu Y (April 2014). "8-oxoguanine causes spontaneous de novo germline mutations in mice". Scientific Reports. 4: 4689. Bibcode:2014NatSR...4E4689O. doi:10.1038/srep04689. PMC 3986730. PMID 24732879.
- ↑ "The causes of mutations". evolution.berkeley.edu. Retrieved 2017-11-30.
- ↑ Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME (February 2016). "Timing, rates and spectra of human germline mutation". Nature Genetics. 48 (2): 126–133. doi:10.1038/ng.3469. PMC 4731925. PMID 26656846.
- ↑ Cai L, Wang P (March 1995). "Induction of a cytogenetic adaptive response in germ cells of irradiated mice with very low-dose rate of chronic gamma-irradiation and its biological influence on radiation-induced DNA or chromosomal damage and cell killing in their male offspring". Mutagenesis. 10 (2): 95–100. doi:10.1093/mutage/10.2.95. PMID 7603336.
- 1 2 3 4 "Mutations and Disease | Understanding Genetics". genetics.thetech.org. Retrieved 2017-11-30.
- 1 2 "The Genetics of Cancer". Cancer.Net. 2012-03-26. Retrieved 2017-12-01.
- 1 2 "The Genetics of Cancer". National Cancer Institute. NIH. 2015-04-22. Retrieved 23 September 2018.
- 1 2 "Huntington disease". Genetics Home Reference. NIH. Retrieved 23 September 2018.
- ↑ Lawrence, David M. (2009). Huntington's Disease. New York, NY 10001: Infobase Publishing. p. 92. ISBN 9780791095867.
{{cite book}}: CS1 maint: location (link) - ↑ "Huntington's disease". Mayo Clinic. Retrieved 23 September 2018.
- 1 2 3 Chandley AC (April 1991). "On the parental origin of de novo mutation in man". Journal of Medical Genetics. 28 (4): 217–23. doi:10.1136/jmg.28.4.217. PMC 1016821. PMID 1677423.
- ↑ Hook, EB (September 1981). "Rates of chromosome abnormalities at different maternal ages". Obstetrics and Gynecology. 27 (1): 282–5. doi:10.1016/0091-2182(82)90145-8. PMID 6455611.
- ↑ Ghanta, Sujana (October 2010). "Non-Invasive Prenatal Detection of Trisomy 21 Using Tandem Single Nucleotide Polymorphisms". PLOS ONE. 5 (10): e13184. Bibcode:2010PLoSO...513184G. doi:10.1371/journal.pone.0013184. PMC 2951898. PMID 20949031.
- 1 2 3 4 "Cystic Fibrosis Canada". www.cysticfibrosis.ca. Retrieved 2017-11-30.
- ↑ O'Sullivan BP, Freedman SD (May 2009). "Cystic fibrosis". Lancet. 373 (9678): 1891–904. doi:10.1016/S0140-6736(09)60327-5. PMID 19403164. S2CID 46011502.
- ↑ Reference, Genetics Home. "CFTR gene". Genetics Home Reference. Retrieved 2017-11-30.
- ↑ "Prenatal Diagnosis". Johns Hopkins Cystic Fibrosis Center. Retrieved 23 September 2018.
- 1 2 3 4 5 6 Sander JD, Joung JK (April 2014). "CRISPR-Cas systems for editing, regulating and targeting genomes". Nature Biotechnology. 32 (4): 347–55. doi:10.1038/nbt.2842. PMC 4022601. PMID 24584096.
- ↑ "About Human Germline Gene Editing | Center for Genetics and Society". www.geneticsandsociety.org. Retrieved 2017-12-01.
- ↑ Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C, Wang Y, Brodsky RA, Zhang K, Cheng L, Ye Z (July 2014). "Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs". Cell Stem Cell. 15 (1): 12–3. doi:10.1016/j.stem.2014.06.011. PMC 4338993. PMID 24996165.
- 1 2 3 Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG, Tan W, Penheiter SG, Ma AC, Leung AY, Fahrenkrug SC, Carlson DF, Voytas DF, Clark KJ, Essner JJ, Ekker SC (November 2012). "In vivo genome editing using a high-efficiency TALEN system". Nature. 491 (7422): 114–8. Bibcode:2012Natur.491..114B. doi:10.1038/nature11537. PMC 3491146. PMID 23000899.
- ↑ Nemudryi AA, Valetdinova KR, Medvedev SP, Zakian SM (July 2014). "TALEN and CRISPR/Cas Genome Editing Systems: Tools of Discovery". Acta Naturae. 6 (3): 19–40. doi:10.32607/20758251-2014-6-3-19-40. PMC 4207558. PMID 25349712.
- 1 2 3 Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (September 2010). "Genome editing with engineered zinc finger nucleases". Nature Reviews Genetics. 11 (9): 636–46. doi:10.1038/nrg2842. PMID 20717154. S2CID 205484701.