Holocarboxylase synthetase deficiency
| Holocarboxylase synthetase deficiency | |
|---|---|
| Other names: Early-onset multiple carboxylase deficiency[1] | |
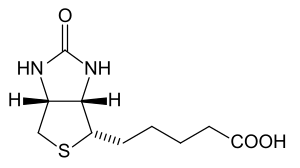 | |
| Biotin | |
Holocarboxylase synthetase deficiency is an inherited metabolic disorder in which the body is unable to use the vitamin biotin effectively.[2] This disorder is classified as a multiple carboxylase deficiency, a group of disorders characterized by impaired activity of certain enzymes that depend on biotin. Symptoms are very similar to biotinidase deficiency and treatment – large doses of biotin – is also the same.
Symptoms and signs
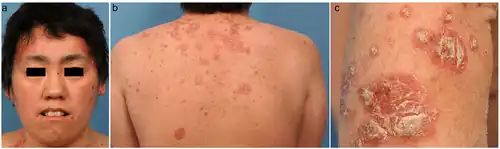
The presentation of Holocarboxylase synthetase deficiency is via the following:[3]
- Exfoliative dermatitis
- Poor appetite
- Vomiting
- Irritability
Genetics
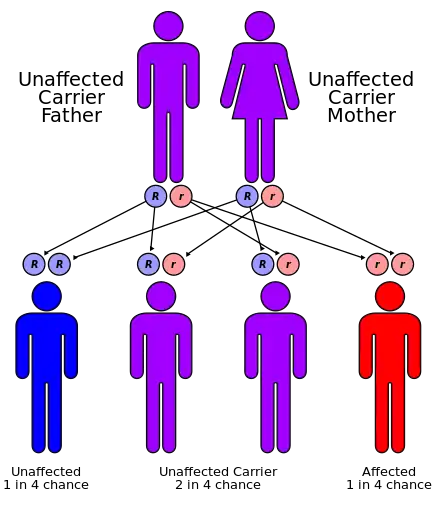
Mutations in the HLCS gene cause holocarboxylase synthetase deficiency. The HLCS gene makes an enzyme, holocarboxylase synthetase, that attaches biotin to other molecules. Biotin, a B vitamin, is found in foods such as liver, egg yolks, and milk. It is essential for the normal production and breakdown of proteins, fats, and carbohydrates in the body. Mutations in the HLCS gene reduce the activity of holocarboxylase synthetase, preventing cells from using biotin effectively and disrupting many cellular functions. This condition is inherited in an autosomal recessive pattern, which means two copies of the gene in each cell are altered.
Diagnosis
The signs and symptoms of holocarboxylase synthetase deficiency typically appear within the first few months of life, but the age of onset varies. Affected infants often have immunodeficiency diseases, difficulty feeding, breathing problems, a skin rash, hair loss (alopecia), and a lack of energy (lethargy). Immediate treatment and lifelong management (using biotin supplements) may prevent many of these complications. If left untreated, the disorder can lead to delayed development, seizures, and coma. These medical problems may be life-threatening in some cases.
Treatment
In terms of treatment we find that for HCSD it is biotin supplementation [3]
See also
References
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Holocarboxylase synthetase deficiency". www.orpha.net. Archived from the original on 29 November 2017. Retrieved 8 April 2019.
- ↑ Reference, Genetics Home. "holocarboxylase synthetase deficiency". Genetics Home Reference. Archived from the original on 2017-05-13. Retrieved 2017-05-09.
- 1 2 RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Holocarboxylase synthetase deficiency". www.orpha.net. Archived from the original on 27 November 2022. Retrieved 20 April 2023.
This article incorporates public domain text from The U.S. National Library of Medicine Archived 2019-02-04 at the Wayback Machine
External links
| Classification | |
|---|---|
| External resources |