Idiopathic generalized epilepsy
| Idiopathic generalized epilepsy | |
|---|---|
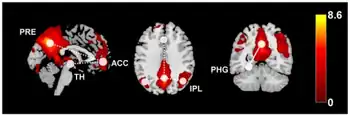 | |
| Diagram summarizes the functional connectivity (FC) changes in individuals with idiopathic generalized epilepsy compared to healthy controls. | |
| Specialty | Neurology |
Idiopathic generalized epilepsy (IGE) is a group of epileptic disorders that are believed to have a strong underlying genetic basis.[1] Patients with an IGE subtype are typically otherwise normal and have no structural brain abnormalities.[2] People also often have a family history of epilepsy and seem to have a genetically predisposed risk of seizures.[1] IGE tends to manifest itself between early childhood and adolescence although it can be eventually diagnosed later.[2] The genetic cause of some IGE types is known, though inheritance does not always follow a simple monogenic mechanism.[1]
IGE is a group of epileptic disorders that are believed to have a strong underlying genetic basis.[3] It contains sporadic mendelian or monogenic epilepsies.[1] A genetic predisposition play a key part behind the reason of the idiopathic generalized epilepsies.[4] Patients with an IGE subtype are typically otherwise normal and have no structural brain abnormalities.[4] People also often have a family history of epilepsy and seem to have a genetically predisposed risk of seizures.[4] IGE tends to manifest itself between early childhood and adolescence although it can be eventually diagnosed later.[4] The genetic cause of some IGE types is known, though inheritance does not always follow a simple monogenic mechanism.[5] Among all epilepsy, Idiopathic generalized epilepsy (IGE) is very common.[5] The clinical characteristics of IGE are absence seizures, myoclonic seizures, and tonic-clonic seizures.[5] The onset of Idiopathic generalized epilepsy (IGE) in early infancy, childhood, and Idiopathic generalized epilepsy (IGE), but it sometimes may start in adulthood.[6] Seizures were categorized as partial, generalized, and unclassified by ILAE.[3] IGE-associated seizures can happen in various degrees and combinations.[3] Plentiful categories of status epilepticus can be seen in IGE.[7] IGE consists of various mixtures of generalized tonic-clonic, absence seizures, and myoclonic jerks.[2]
Types
Benign myoclonic epilepsy in infancy (BMEI)
This form of epilepsy is very rare, and is twice as prevalent in boys compared to girls.[6] It representing less than 2% of cases in IGE and 1% of all the epilepsy.[8] Age of seizure onset is between 4 months and 3 years of age.[6] Children with this disorder often present with head drops and brief arm jerks.[6] Although there is believed to be a genetic basis for this disorder, no genetic linkage has been shown.[8] There are few findings related to similar family history and also evidence of autosomal dominance in few cases.[8] There are few linkages in the chromosomal zone but there was no precise gene has been found.[8] There was evidence of febrile seizure has been found almost in 50% of the cases.[6] Some times at the initials stage it was misdiagnosed (differential diagnose) as West syndrome.[6] The first line of treatment for BMEI is anti seizure medication that is Valproic acid (VPA).[6] when the seizure could not be controlled by Valproic acid (VPA) then lobazam (CLB) or ethosuximide (ESM) can be used as replacements.[6]
Generalized epilepsy with febrile seizures plus
Generalized epilepsy with febrile seizures plus (GEFS+) is an umbrella for many other syndromes that share causative genes.[9] Patients experience febrile seizures early in childhood and grow to experience other types of seizures later in life.[10] In GEFS+ febrile seizures started at the age of 6 month and it will continue till more than 6 years of age.[11] During the period, sometimes fever is present sometimes fever was absent.[11] Known causative genes for GEFS+ are the sodium channel α subunit genes SCN1A and SCN2A and the β subunit gene SCN1B. Mutations in the GABAA receptor γ subunit GABRG1 are also causative for this disorder.[11] Sometimes it’s comorbid with Autism, anxiety, and mental retardation.[11] GEFS+ is inherited and one of the causal factor is autosomal dominance.[12] The treatment for GEFS+ depend on the severity and type of seizures.[10] The children who are having the more complex forms of seizures need a combination of antiepileptic drugs.[10] The children who are having febrile convulsion do not require complex antiepileptic medicines.[10] as it is a rare entity, there isn't much evidence for the actual prevalence.[9] The diagnosis of GEFS+ mostly based on the clinical history, family history and neurological evaluations.[10] As mutation is one foe the major reason for GEFS+, so blood test for finding out the mutation could be one of the diagnostic process.[10]
Epilepsy with myoclonic absences (EMA)
This rare epilepsy has a wide age range of presentation (from the first year of life through the early teens).[10] This syndrome can be found in 1 in 100-200 children with epilepsy.[10] In comparison to the girls it is mostly found in boys.[13] This epilepsy is characterized by absence seizures concurrent with myoclonic jerks, typically occurring several times daily.[13] There is no evidence of focal seizures, and also absence seizures concurrent with myoclonic jerks.20. [14] The genetics of this disorder have not been delineated,[13] but few case studies showed that there is a relation with the SYNGAP1 gene.[14] Seizures from this disorder often cease within 5 years.[13] Diagnosis of EMA is basically based on clinical features and clinical history.[10] EEG plays a major role in the diagnosis of EMA.[10] Abnormal EEG can be recorded due to hyperventilation.[14] Sometimes for diagnosis, the clinicians tried to trigger the seizure by getting the child to over-breathe.[10] MRI is required to find out the structural changes in the brain.[14] Though the imaging is normal, there were few atrophy can be seen.[14] The seizures were not well controlled by monotherapy.[13] Combination of several anti-epileptic drug is useful for the treatment of seizures in AME.[13] Lamotrigine and sodium valproate, or ethosuximide and sodium valproate, combined are pretty useful and effective.[10] Clinicians should avoid the medications such as carbamazepine, oxcarbazepine, eslicarbazepine and phenytoin.[13] Surgery is not as useful in this case.[13]
Epilepsy with myoclonic-astatic seizures
Originally called Doose syndrome, epilepsy with myoclonic-astatic seizures accounts for ~2% of childhood epilepsies. Children with this disorder have incredibly brief (<100ms) myoclonic jerks followed by equally brief loss of muscle tone, sometimes resulting in dangerous falls. Some patients have much longer lasting seizures of this type. Many patients with this disorder also have absence seizures. This is believed to be a polygenic disorder.
Childhood absence epilepsy
Also known as pyknolepsy, childhood absence epilepsy (CAE) represents up to 10% of all childhood epilepsies.[15] It first manifests in childhood between the ages of 2 and 12 as brief periods of unconsciousness (absence).[15] It can ben found equally in both males and females.[15] Mutations in the calcium channel α subunit encoding gene CACNA1H and the GABA receptor γ subunit encoding gene GABRG2 yield susceptibility for CAE.[16] Patient with CAE might have the history of febrile seizure.[15] Though the cognition and developmental milestones are normal, in some cases Attention deficit hyperactivity disorder and learning disabilities can be comorbid with CAE.[15] They can interfere with children's academic performances and daily lives.[17] The occurrence absence seizures vary from 0.7 to 4.6/100,000 in overall population and 6 to 8/100,000 in children up to 15 years-old.[18]
Juvenile absence epilepsy
Juvenile absence epilepsy is similar to CAE but has an onset between ages 9 and 12, sometimes it may start earlier at the age of 6–7 years[10] In JAE the child may experience both absence seizures as well as tonic-clonic seizures.[19] The duration of seizures lasts for 5-20 sec or in some cases maximum 40 sec.[10] The patients with this disorder have less frequent but longer absence seizures than those with CAE.[19] Though the cognition is intact, but attention deficit hyperactivity disorder and learning difficulties sometimes comorbid with JAE.[14] Roughly 1 to 2 in 100 case with epilepsy have been diagnosed as JAE.[20] Diagnosis of JAE is mainly on the basis of seizure type, clinical history, family history, and physical and neurological examinations by expert clinicians.[10] EEG and MRI have been necessary to perform just to find out the seizure activity and whether any lesion involves, though the MRI has been found normal in patients with JAE.[20] There are a number of possible genetic loci for this disorder, though no causative genes have been demonstrated.[20] The useful anti-seizure medication for JAE are ethosuximide (Zarontin), sodium valproate (Epilim) and lamotrigine (Lamictal).[10][20] Ketogenic diet is also an treatment option and also do not have side effects.[10]
Juvenile myoclonic epilepsy
Also known as Janz syndrome,[21] juvenile myoclonic epilepsy (JME) is a common form of epilepsy, accounting for ~10% of all cases and ~25% of cases of idiopathic generalized epilepsies.[22] Many children with CAE go on to develop JME.[22] JME first presents between the ages of 12 and 18 with prominent myoclonic seizures.[22] These seizures tend to occur early in the morning.[22] Patients with JME may also have generalized tonic-clonic seizures and absence seizures.[22] Linkage of this disorder has been shown to mutations in the genes GABRA1, CACNB4, CLCN2, GABRD2, EFHC1, and EFHC2.[21][23] The onset of Juvenile myoclonic epilepsy is in adolescence, and it might continued lifelong.[21]
Epilepsy with generalized tonic-clonic seizures only
This type of IGE can present at almost any age and is poorly characterized. Because of its loose definition, it is impossible to supply an accurate estimate of its prevalence. As implied by its name, patients with this disorder present only with tonic-clonic seizures.
Symptoms and signs
Before the seizure begins-[24]
- Odd changes in taste
- Changes in emotions
- Changes in Vision
- Changes in Smell[24]
During seizure
- bite your cheek or tongue
- lock your jaw
- lose control of your bladder or bowels
- turn blue in the face[24]
After the seizure[24]
- No Memory event
- Drowsiness
- Headache
- Confusion
Cognitive Function
The cognitive problem can be seen in 40%-60% of IGE patients.[25] Patients with uninhibited epilepsy are more prone to have cognitive dysfunctions.[25] Polypharmacy pharmacological treatment is one of the factors for cognitive dysfunction in IGE.[25]
Causes
The particular causal factor leading to IGE is unknown, but the primary cause for IGE found to be genetic.[26] There are specific mutations and chromosomal abnormalities noted in IGE.[16] There is evidence from different studies of the association between IGE and chromosome 15.[26] Irregular GABA receptor function is one of the causal factors for IGE.[16] As the main cause of the IGE is genetic, it can be inherited from parents due to some genetic modifications.[26] IGE is related to different gene mutation, which is either new or inherited.[16] The genetic mutations that cause IGE are not completely clear, but it is not a single gene mutation.[16] one of the genes that is responsible for IGE is CHRNA7.[26] The genetic causal evidence can be seen in a different study conducted in monozygotic twins than dizygotic twins.[27]
Diagnosis
Although the diagnosis of IGE is primarily based on clinical features, electroencephalography (EEG) play an vital role in diagnosis of IGE.[28] Sleep deprivation is one of the cause for having abnormal EEG pattern.[16] There was no abnormality found in the interictal EEG.[6] In a few cases, discharge of spike waves can be seen in both awake and sleeping state.[6] Diagnosis is based on the clinical history, family history, physical and neurological examination done by the experts.[16] Study shows that there are no significant structural changes were found in MRI of IGE patients.[27]
Treatment
Management for Idiopathic generalized epilepsy is based on antiepileptic medication[29]
Epidemiology
The worldwide prevalence for IGE is 3 6/1000.[8] The prevalence of IEG varies from study to study.[3] In some, it is as low as, and in some, it is as high as.[3] It is found to be high in those study that includes children and adolescents with epilepsy.[3] The onset of IGE is mostly found in childhood to adolescence, but in few cases, the onset has been found after 60 years of age.[30] The late onset of IGE is mostly found in women.[30]
References
- 1 2 3 4 Gardiner, M (2005). "Genetics of idiopathic generalized epilepsies". Epilepsia. 46 Suppl 9: 15–20. doi:10.1111/j.1528-1167.2005.00310.x. PMID 16302872. S2CID 31002006.
- 1 2 3 Panayiotopoulos, Chrysostomos P. (November 2005). "Idiopathic Generalized Epilepsies: A Review and Modern Approach". Epilepsia. 46 (s9): 1–6. doi:10.1111/j.1528-1167.2005.00330.x. PMID 16302869. S2CID 45688336.
- 1 2 3 4 5 6 Beydoun, A; D'Souza, J (June 2012). "Treatment of idiopathic generalized epilepsy - a review of the evidence". Expert Opinion on Pharmacotherapy. 13 (9): 1283–98. doi:10.1517/14656566.2012.685162. PMID 22568658. S2CID 207479596.
- 1 2 3 4 Sander, T (April 1996). "The genetics of idiopathic generalized "epilepsy: implications for the understanding of its aetiology". Molecular Medicine Today. 2 (4): 173–80. doi:10.1016/1357-4310(96)88793-4. PMID 8796880.
- 1 2 3 Marini, C; King, MA; Archer, JS; Newton, MR; Berkovic, SF (February 2003). "Idiopathic generalised epilepsy of adult onset: clinical syndromes and genetics". Journal of Neurology, Neurosurgery, and Psychiatry. 74 (2): 192–6. doi:10.1136/jnnp.74.2.192. PMC 1738270. PMID 12531947.
- 1 2 3 4 5 6 7 8 9 10 Caraballo, RH; Dalla Bernardina, B (2013). "Idiopathic generalized epilepsies". Handbook of Clinical Neurology. 111: 579–89. doi:10.1016/B978-0-444-52891-9.00060-9. ISBN 9780444528919. PMID 23622205.
- ↑ Shorvon, S; Walker, M (2005). "Status epilepticus in idiopathic generalized epilepsy". Epilepsia. 46 Suppl 9: 73–9. doi:10.1111/j.1528-1167.2005.00316.x. PMID 16302878. S2CID 26086785.
- 1 2 3 4 5 Prasad, DK; Satyanarayana, U; Munshi, A (November 2013). "Genetics of idiopathic generalized epilepsy: an overview". Neurology India. 61 (6): 572–7. doi:10.4103/0028-3886.125240. PMID 24441321.
- 1 2 "Genetic epilepsy with febrile seizures plus: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 2022-08-15. Retrieved 2022-08-23.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 "GEFS+ Genetic epilepsy with febrile seizures plus | Epilepsy Action". www.epilepsy.org.uk. Archived from the original on 2022-09-30. Retrieved 2022-08-23.
- 1 2 3 4 Tan, EH; Yusoff, AA; Abdullah, JM; Razak, SA (May 2012). "Generalized epilepsy with febrile seizure plus (GEFS+) spectrum: Novel de novo mutation of SCN1A detected in a Malaysian patient". Journal of Pediatric Neurosciences. 7 (2): 123–5. doi:10.4103/1817-1745.102575. PMC 3519070. PMID 23248692.
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Generalized epilepsy with febrile seizures plus". www.orpha.net. Archived from the original on 2022-08-15. Retrieved 2022-08-23.
- 1 2 3 4 5 6 7 8 "Epilepsy Myoclonic Absences". Epilepsy Foundation. Archived from the original on 2022-08-16. Retrieved 2022-08-23.
- 1 2 3 4 5 6 "EPILEPSY WITH MYOCLONIC ABSENCES". www.epilepsydiagnosis.org. Archived from the original on 2022-08-17. Retrieved 2022-08-23.
- 1 2 3 4 5 "CHILDHOOD ABSENCE EPILEPSY". www.epilepsydiagnosis.org. Archived from the original on 2022-08-11. Retrieved 2022-08-23.
- 1 2 3 4 5 6 7 "Idiopathic Generalized Epilepsy: An Overview | MyEpilepsyTeam". www.myepilepsyteam.com. Archived from the original on 2022-08-09. Retrieved 2022-08-23.
- ↑ Khosravani, H; Bladen, C; Parker, DB; Snutch, TP; McRory, JE; Zamponi, GW (May 2005). "Effects of Cav3.2 channel mutations linked to idiopathic generalized epilepsy". Annals of Neurology. 57 (5): 745–9. doi:10.1002/ana.20458. PMID 15852375. S2CID 28752058.
- ↑ Guilhoto, LM (January 2017). "Absence epilepsy: Continuum of clinical presentation and epigenetics?". Seizure. 44: 53–57. doi:10.1016/j.seizure.2016.11.031. PMID 27986418. S2CID 205140359.
- 1 2 "Juvenile Absence Epilepsy (JAE)| Children's Hospital Pittsburgh". Children's Hospital of Pittsburgh. Archived from the original on 2021-10-20. Retrieved 2022-08-23.
- 1 2 3 4 "Juvenile Absence Epilepsy". Epilepsy Foundation. Archived from the original on 2022-09-02. Retrieved 2022-08-23.
- 1 2 3 Gilsoul, M; Grisar, T; Delgado-Escueta, AV; de Nijs, L; Lakaye, B (2019). "Subtle Brain Developmental Abnormalities in the Pathogenesis of Juvenile Myoclonic Epilepsy". Frontiers in Cellular Neuroscience. 13: 433. doi:10.3389/fncel.2019.00433. PMC 6776584. PMID 31611775.
- 1 2 3 4 5 Caciagli, L; Wandschneider, B; Xiao, F; Vollmar, C; Centeno, M; Vos, SB; Trimmel, K; Sidhu, MK; Thompson, PJ; Winston, GP; Duncan, JS; Koepp, MJ (1 September 2019). "Abnormal hippocampal structure and function in juvenile myoclonic epilepsy and unaffected siblings". Brain: A Journal of Neurology. 142 (9): 2670–2687. doi:10.1093/brain/awz215. PMC 6776114. PMID 31365054.
- ↑ Santos, BPD; Marinho, CRM; Marques, TEBS; Angelo, LKG; Malta, MVDS; Duzzioni, M; Castro, OW; Leite, JP; Barbosa, FT; Gitaí, DLG (2017). "Genetic susceptibility in Juvenile Myoclonic Epilepsy: Systematic review of genetic association studies". PLOS ONE. 12 (6): e0179629. Bibcode:2017PLoSO..1279629S. doi:10.1371/journal.pone.0179629. PMC 5479548. PMID 28636645.
- 1 2 3 4 "Epilepsy with Generalized Seizures: Symptoms, Causes, and Treatments". Healthline. 20 July 2012. Archived from the original on 30 September 2022. Retrieved 23 August 2022.
- 1 2 3 Koganti, H; Paneyala, S; Sundaramurthy, H; Sc, N; Kashyap, RS; Joshi, S; Colaco, V (July 2020). "The Impact of Idiopathic Generalized Epilepsy on Executive Functions". Annals of Neurosciences. 27 (3–4): 131–135. doi:10.1177/0972753120968751. PMC 8454999. PMID 34556951.
- 1 2 3 4 "Idiopathic Epilepsy: Symptoms, Causes and Treatment". Life Persona. Archived from the original on 2022-05-11. Retrieved 2022-08-23.
- 1 2 McWilliam, M; Al Khalili, Y (January 2022). "Idiopathic (Genetic) Generalized Epilepsy". PMID 31536218.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Seneviratne, U; Cook, M; D'Souza, W (February 2012). "The electroencephalogram of idiopathic generalized epilepsy". Epilepsia. 53 (2): 234–48. doi:10.1111/j.1528-1167.2011.03344.x. PMID 22150583. S2CID 6127915.
- ↑ McWilliam, Matthew; Al Khalili, Yasir (2022). "Idiopathic (Genetic) Generalized Epilepsy". StatPearls. StatPearls Publishing. Archived from the original on 2 September 2022. Retrieved 29 September 2022.
- 1 2 Loiseau, J; Crespel, A; Picot, MC; Duché, B; Ayrivié, N; Jallon, P; Loiseau, P (December 1998). "Idiopathic generalized epilepsy of late onset". Seizure. 7 (6): 485–7. doi:10.1016/s1059-1311(98)80007-1. PMID 9888493. S2CID 25126529.
Sources
- "Proposal for Revised Classification of Epilepsies and Epileptic Syndromes". Epilepsia. 30 (4): 389–99. 1989. doi:10.1111/j.1528-1157.1989.tb05316.x. PMID 2502382. S2CID 3483250.
- Jr, N.; Douglas, R. (2005). "Idiopathic Generalized Epilepsies Recognized by the International League Against Epilepsy". Epilepsia. 46: 48–56. doi:10.1111/j.1528-1167.2005.00313.x. PMID 16302875. S2CID 25513807.