Leukoencephalopathy with neuroaxonal spheroids
Leukoencephalopathy with neuroaxonal spheroids (LENAS) is an extremely rare kind of leukoencephalopathy and is classified as a neurodegenerative disease. LENAS is a cause of severe and subacute dementia that results from damage to certain areas of the brain. This damage is to a type of brain tissue called white matter and axon damage due to swellings which are termed spheroids.[1]
The rarity and unknown prevalence of this disease may be due to most symptoms being similar to other common disorders, leading to misdiagnosis.[2] LENAS normally has an adult onset (but also can be present in childhood), which can present on MRIs that mimics progressive multiple sclerosis and is thus misdiagnosed for this instead.[3]
The genetic etiology of LENAS is known to follow an autosomal dominant pattern through a mutation in the CSF1R gene.
Signs and Symptoms
The signs and symptoms that are present vary with each individual as some may have all symptoms while some may only have some listed below. However, the progression of this disease is different with each individual which reflects how the symptoms change over time. Further, the damage that we see to the myelin and axons is thought to contribute to many of the neurological signs and symptoms that are shown in this condition.[1] The most frequent symptoms are categorized into psychiatric, psychotic and neurologic.[4]
| Category | Symptoms |
|---|---|
| Psychiatric | Depression, Anxiety, Alcohol use disorder, Irritability, and Aggressiveness |
| Psychotic | Confusion, Delusions, and Hallucinations |
| Neurologic | Dementia, Seizures, Impaired Balance, Retropulsion, Gait Apraxia, Spasticity, Ataxia, and Urinary Incontinence |
General Symptoms
- Personality Changes[1]
- Loss of social inhibitions[1]
- Depression[1]
- Memory Loss[1]
- Loss of Executive Function[1]
- The ability to plan and use problem-solving skills inhibits normal life skills such as impulse control and appropriately paying attention in general[1]
- Seizures[1]
- Severe decline in thinking and reasoning abilities (dementia ).[1]
- Motor Skills become affected, many may have difficulty walking[1]
- Patterns of movement abnormalities can occur as well such as:[1]
- Parkinsonism :[1]
- Generally will see bradykinesia , tremors, and muscle rigidity.[1]
- Parkinsonism :[1]
- Patterns of movement abnormalities can occur as well such as:[1]
While these vary among individuals as mentioned, over time, almost all patients with this condition will be unable to walk, speak and care for themselves.[1]
Causes
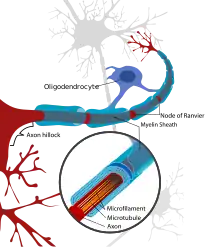
Leukoencephalopathy with neuroaxonal spheroids (LENAS) is believed to occur when presence of white matter degeneration and axonal spheroids are seen with a brain biopsy or an MRI.[5][2]
White matter consists of nerve fibers (axons) covered by a substance called myelin that insulates and protects them.[2] The axons extend from nerve cells (neurons) and transmit nerve impulses throughout the body.[2]
The result of spheroids in the brain leads to this significant decline of functioning that progressively worsens the brain function and leads to varied symptoms. However, why this occurs is still not entirely understood, thus more research is continuously being done to this day.
There have been identifiable genetic differences that are better understood. This disease is inherited while following an autosomal dominant pattern.
Genetic Etiology
There are identifiable genetic causes that are better understood with this disease. LENAS is caused by a mutation in a gene, specifically the CSF1R gene. This mutation changes protein receptors on the gene which normally plays a role in important cell signaling pathways; however, this altercation inhibits the regular function.[1]
Normal Function of CSF1R Gene
The colony stimulating factor 1 receptor (CSFR1) gene regularly functions by giving instructions for making a protein termed the colony stimulating factor 1 receptor (CSF-1 receptor).[6] Proteins in general attach (bind) to their specific receptor which "turns on" (activates) to stimulate a cascade of cellular signaling pathways crucial for cell function to occur. These events occur with the specific CSF-1 protein. As it binds and is activated, it allows for important cellular processes to occur which include cell growth, division, and maturation of cells to in turn take on specific functions.[6]
Glial cells, located in the brain, are responsible for protection and maintenance of neurons. In a healthy brain, the membrane of glial cells are abundant with the CSF-1 receptor gene and is also thought to be an important player in the proliferation and differentiation of these cells.[6]
| Leukoencephalopathy with neuroaxonal spheroids | |
|---|---|
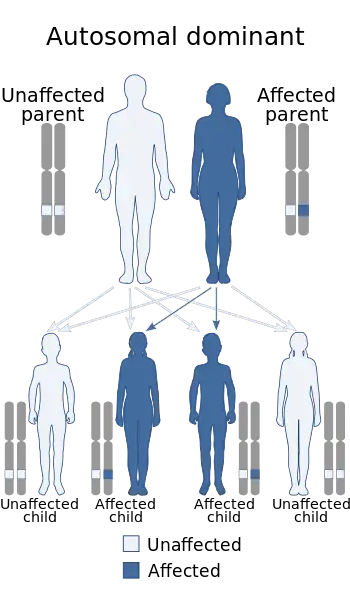 | |
| This condition is inherited in an autosomal dominant manner | |
| Specialty | Neurology |
Mutated CSF1R Gene
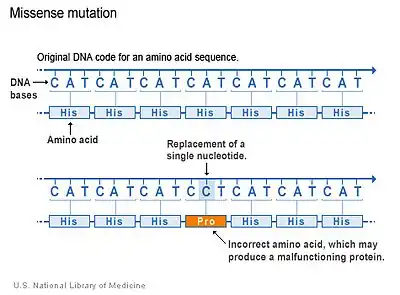
There are several types of mutations that occur in genes. The majority of CSF1R genetic mutations in LENAS occur due to a type of mutation which is called a missense mutation.[2] Missense mutations occur when there is a change in a single amino acid of a protein. This single change can result in problems with functions of the protein, like the CSF-1 receptor. The kinase domain, which is the region of CSF-1 receptor where the mutation occurs, is altered and thus the normal function that activates other proteins is compromised and cannot stimulate cell signaling pathways properly.[2]
Although this is the primary type of mutation in LENAS, other types may occur as well but it is not as well understood for this disease.[2]
This genetic mutation is related to LENAS and said to be the main cause but how these lead to damage in the white matter and associated symptoms (cognitive and movement impairment symptoms) is still not fully clear.[2][6]
Inheritance
LENAS is inherited in an autosomal dominant pattern. This means that one copy of the mutated gene in each cell is sufficient to cause the disorder.[1]
If one parent who is unaffected and one parent who is affected with LENAS give birth to four children, at least two of the four will become affected and have the disorder because the affected parent would pass on the one copy of the gene. In most cases, an affected person does inherit the mutation from one affected parent but not all cases.
There have been very few reports of cases that result from new mutations have been seen as well as cases in which there has been no history of LENAS.[1] These few reports are not as concrete or well understood yet and further studies should be conducted.
Pathophysiology
Neuropathology
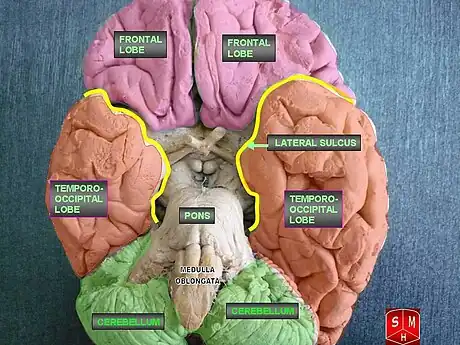
LENAS is seen with damage to the white matter and axons within the brain. The external human LENAS brain shows findings in several major structures. There is mild atrophy of the frontoparietal regions of the brain and a mild reduction of the thalamus and rostral (front) part of the caudate nucleus (which is located in an area of the brain called the basal ganglia).[4] Abnormalities in the frontal, frontoparietal, and temporal lobes are most severe and predominant with LENAS and asymmetry of the cerebral hemispheres has sometimes been found.[4] LENAS also may show moderately enlarged lateral ventricles and atrophy in corticospinal tracts as well as in the pons.[4]
The area where it is seen to be the most pronounced abnormalities appear in the white matter below the pre- and postcentral gyri that extend through the posterior limb of the internal capsule into pyramidal tracts of the brain stem.[4]
- The corpus callosum is variably affected.
- Reactive astrocytes and macrophages are present, but no inflammation appears.
- The cerebral cortex and basal ganglia are normal and contain no or only few spheroids.
- Within the cerebellum, there is a marked loss of Purkinje cells seen but cerebral white matter is normal.
Specific Immunostains are used as the easiest identification of neuroaxonal spheroids in LENAS which appear as round to oval shaped swellings and are seen in affected white matter.[4] If there appears to be a large amount of loss in myelin sheaths on axons and these spheroids, LENAS progressed to become widespread.[4]
Electron Microscopy has also been used to identify spheroids. In LENAS, evidence of the spheroids may show neurofilaments that are scattered amount electron-dense material and mitochondria.[4]
Subcortical U-fibers in the brain appear to be relatively spared, meaning they seem to not be involved in most cases but this does not mean they are not always/eventually involved.[4][7] U-fibers represent connections which are between adjacent areas of the brain located within the cortex or deep in the white matter that are one of the last parts of the brain to be myelinated.[7] In LENAS, it has been found that because these U-fibers are last to myelinate normally, they are also last to be affected as the disease progresses.
Biochemistry
The primary biochemical defect in LENAS disrupts normal states and may involve oxidative stress. Ceroids, which are essentially products of unsaturated fatty acids that build up, are found in macrophages and other glia is thought to be an end-product of oxidative damage, indicative of membrane damage from abnormal accumulation.[8]
The high levels of Iron is also present in LENAS which can be associated to cause an increased level of toxic free radicals and causing oxidative damage to the body, causing failure in our cellular energy systems.[9]
Disease Mechanism
The mechanism of LENAS still seems to remain unclear and it varies as more research on this is being done. Some mechanisms that can be of clinical importance of why individuals show certain symptoms and which area of the brain has been shown to be consistent.[9]
Normally, the frontal lobe in our brain are important for our higher level executive functions. Predominance of white matter damage in this lobe has been found to be consistent with both the psychiatric and behavioral signs and symptoms pertaining to LENAS.[9] The underlying symptoms seen of frontotemporal dementia seen commonly in LENAS is associated with the temporal lobe damage.[9] There is a large disconnect in the brain between the lobes that reflect the neuropsychiatric symptoms that are common with the disease.[9]
Ataxia, which is related to our daily voluntary movements of muscles, are often present even in patients without cerebellar involvement which could reflect either minimal damage to the cerebellum or diffuse cerebral white matter lesions.[9]
Diagnosis
The diagnosis of LENAS is usually based on medical and family history, genetic testing, extensive imaging and other supplemental testing. As this disease is extremely rare, diagnosis is still very complex as many of these diagnostic criteria for LENAS can be mistaken for similar neurodegenerative disorders. The differential diagnosis of leukoencephalopathy in general is very extensive and specialized investigations are required to make an accurate diagnosis.[10]
Diagnostic Criteria
Note: These criteria were established by a group consisting of board-certified neurologists from the Mayo Clinic, Niigata University, Shinshu University School of Medicine, Kyoto Prefectural University of Medicine, and Tokushima University Graduate School.
.jpg.webp)
Core Features
- Age at onset ≤ 60 years old[10]
- More than 2 findings of the following clinical signs and symptoms:[10]
- Autosomal Dominant inheritance or sporadic occurrence[10]
- Brain CT/MRI findings:
- Other causes of Leukoencephalopathy including vascular dementia, multiple sclerosis, or leukodystrophy can be excluded.[10]
Exclusionary Findings
- Age at onset ≤ 10 years[10]
- Stroke-like episodes more than twice except for epilepsy[10]
- Prominent peripheral neuropathy[10]
Supportive Findings
- Frontal lobe dysfunction shown by clinical features or cognitive battery test[10]
- Rapidly progressive course. Become bedridden within 5 years after onset[10]
- Spotty small calcifications in the white matter shown by brain CT[10]
- Neuropathologic findings compatible to LENAS[10]
| Definite | Probable | Possible |
|---|---|---|
| Fulfills core features 2,3 and 4A and confirmation of CSF1R mutation[10] | Fulfills core features 1-5, but genetic testing has not been performed[10] | Fulfills core features 2a, 3, and 4a, but genetic testing has not been performed[10] |
Genetic Testing
Single-Gene testing is first performed for sequence analysis of the CSF1R gene.[11]
A multi-gene panel that includes CSF1R gene and other genes of interest may be considered for differential diagnoses.[11]
If necessary, more comprehensive genomic testing (not always available though) can be performed.[11]
Further Evaluations
- Complete neurologic assessment[11]
- Psychological and psychiatric assessments[11]
- Assessment of feeding/eating, digestive problems, and nutrition based on clinical history[11]
- EEG if seizure disorder suspected[11]
- Assessment of family and social structure to determine the availability of adequate support systems[11]
- Consultation with a clinical geneticist and/or genetic counselor[11]
Prevention
There currently is not any suggestive findings of preventative measures that should be taken for LENAS before diagnosis. However, prevention of secondary complications can be taken once diagnosis is confirmed.
Prevention of Secondary Complications
Social problems such as unemployment, divorce, financial troubles, and alcoholism as well as suicidal tendencies are often associated as this disease worsens over time. Some of these social consequences may be avoided if family members are informed early about the nature of this disorder as well as if it is diagnosed early enough.[11]
As depression is a major symptom associated, suicidal tendencies may occur. Antidepressant medications can be prescribed for this depression for the attempt to help with depression but they have not shown long-term benefit to date.[11]
Agents and Circumstances to Avoid
The following should be avoided not to prevent the disease, but rather to prevent this disease from progressing faster and causing symptoms to worsen:
- Use of first-generation neuroleptics: these increase seizure risk and risk of additional parkinsonian signs.[11]
- Treatment agents for multiple sclerosis: these have no benefit and have major side effects.[11]
Treatment
No specific therapy for LENAS is currently known or proven to cure this disease, but management of it should be immediately initiated following diagnosis. While more research should be taken, some suggest that hematopoietic stem cell transplantation may show a therapeutic role for this disease.[11]
Management
Management is important in support of this disease and includes attention to general care and nutrition requirements and other possible drug therapies that may help or slow the progression of the disease.[11]
L-dopa or other dopaminergic therapies have not yet been beneficial in individuals with this disease but was noted that it may be worth trying as it does not show negative effects.[11]
Antipsychotics are in general not recommended due to extrapyramidal side effects. However, they may be used in aggressive individuals.[11]
Anti-seizure medications should be initiated in individuals who are having seizure activity along with having this disease as it is reported to be beneficial.[11]
Surveillance
Periodic clinical evaluation and surveillance to monitor the progression of the disease is appropriate to determine if changes need to be made:
- Changes in mobility, communication, and behavior, which could indicate the need to alter care and support systems (for example, moving to a nursing facility or getting personal care, access to wheelchair or walker, etc).[11]
- Onset of seizure activity may cue in the need for anti-seizure therapy[11]
- Contractures, which could indicate the need to change medical management and physical therapy[11]
- More severe behavioral changes that appeared to have worsened. This could include inappropriate emotions and actions, problems following directions, memory loss, and incontinence all which indicate curtailing of independence[11]
- Difficulties in swallowing or weight loss, which trigger physicians consideration for gastrostomy[11]
- Need for physical therapy to minimize contractures and maintain locomotion[11]
Prognosis
Because of how rare this genetic condition is an exact prognosis is still not known and varies. Some data has suggested that the median age of onset is 45 years old, but patients with onset as young as 18 years old has also been described.[5] Further, it is believed that the median life expectancy is 6 years but is also extremely variable as it has also been reported that some patients survived up to 29 years after the onset of symptoms.[5]
While gaining a definitive prognosis for this disease studies suggest a few different techniques that may or may not benefit in getting a better prognosis for the individual patient:
- Lumbar puncture to measure neurofilament light protein (NFL) in the cerebrospinal fluid (CSF) to follow the progression of the disease[11]. An increase NFL level on CSF examinations may suggest faster disease course as well as a worse prognosis.[11]
- Longitudinal MRI studies annually can potentially also help with prognosis. This was found from another study which found that, as throughout the disease course, the more rapid the confluence of patchy or focal T2-weighted hyperintensities and the progression of cortical atrophy suggests the poorer the prognosis appears to be.[12][13][11]
Epidemiology
Epidemiological studies for rare diseases are difficult to have exact and known values that are still difficult to find. However, most studies show the mean age of onset was 43 years old (range 18–78 years old), with a mean death at 53 years (range 23–84 years) and the mean disease duration was found to be 6.8 years (range 1–29 years).[14]
History
LENAS was first reported in multiple members of a large Swedish pedigree in 1984. In this family, 17 out of 71 subjects from 4 generations were affected with this disease.[4] It was found that the age onset of this family varied from 8 to 60 years of age.[4] The age of death in this family was 39 to 89 years with a time between onset and death varied from 3 months to over 30 years.[4] Some patients in this family rapidly developed severe dementia and died a few months after this onset whereas others had a more prolonged progression of the disease.[4] This family was also to have reported sporadic patients as well.[4]
Prevalence
To date, the prevalence of this disease is unclear. Literature has few publications to date of this due to the rarity of the disease. However, a study done in 2011 found information dating back to 1970 that identified 51 individual cases fulfilling criteria to be identified.[15] Today, there could be more confirmed cases but these data have not increased significantly.
Research
The need for future research in this disease is necessary for varied reasons. First, this disease is still so rare and it is to this date very difficult to diagnose. Further, the signs and symptoms of this disease are often confused with other diseases that are more well known and misdiagnosis often occurs. This disease can commonly be mistaken for Multiple Sclerosis (MS) or another form of a more common leukoencephalopathy or neurodegenerative diseases as well. However, some current research has been done primarily on case studies in which scientists are attempting to find new mutations or new ways to diagnose. There are a few studies however that pose to lean in the direction of therapeutical's using hematopoietic stem cell transplantation therapy.
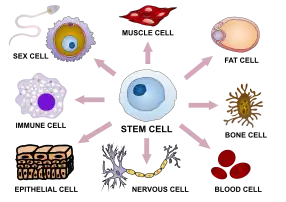
Hematopoietic Stem Cell Therapy
Bone marrow is the soft spongy area in some larger bones of the body that produces many cells which make up red blood cells, white blood cells and platelets.[16] These cells are developed from a type of cell found in bone marrow, termed hematopoietic stem cells.[16] The body is able to direct these stem cells to develop in the blood at any given moment and this is a rapid process.[16] Most of the stem cells remain in the marrow until they are mature which then they are released for specific functions in the body such as carrying oxygen, providing infection protection, and helping blood clotting.[16] Stem cells found in circulating blood are able to be extracted for stem cell therapy use and research.[16]
A study done using hematopoietic stem cell therapy (HSCT) showed clinical benefit but suggested further exploration must be done.[17] Findings using HSCT was beneficial in recessive disorders and saw that it may similarly enhance CSF1R signaling after partial loss seen in LENAS.[17] In the subjects who had LENAS were introduced to HSCT and the finding of further progressed cells was minimal 15 years after the therapy was finished.[17] The most important finding in the subject was that they retained a high level of communication and survived beyond 15 years after onset of symptoms.[17] This is very rare for LENAS as it has been reported to be averaged at 6.8 years for surviving after the onset. This finding provides hope for future research direction and suggests that there may be great benefit to slow the progression of LENAS using HSCT.
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 "Hereditary diffuse leukoencephalopathy with spheroids | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2020-11-10.
- 1 2 3 4 5 6 7 8 "Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: MedlinePlus Genetics". medlineplus.gov. Retrieved 2020-11-10.
- ↑ Keegan BM, Giannini C, Parisi JE, Lucchinetti CF, Boeve BF, Josephs KA (March 2008). "Sporadic adult-onset leukoencephalopathy with neuroaxonal spheroids mimicking cerebral MS". Neurology. 70 (13 Pt 2): 1128–33. doi:10.1212/01.wnl.0000304045.99153.8f. PMID 18287567. S2CID 21341310.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 "Hereditary Diffuse Leukoencephalopathy with Spheroids (HDLS)". United Leukodystrophy Foundation. Retrieved 2020-11-10.
- 1 2 3 Lynch, David S.; Jaunmuktane, Zane; Sheerin, Una-Marie; Phadke, Rahul; Brandner, Sebastian; Milonas, Ionnis; Dean, Andrew; Bajaj, Nin; McNicholas, Nuala; Costello, Daniel; Cronin, Simon (2016-05-01). "Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series". Journal of Neurology, Neurosurgery & Psychiatry. 87 (5): 512–519. doi:10.1136/jnnp-2015-310788. ISSN 0022-3050. PMC 4853550. PMID 25935893.
- 1 2 3 4 "CSF1R gene: MedlinePlus Genetics". medlineplus.gov. Retrieved 2020-11-10.
- 1 2 Gaillard, Frank. "Subcortical U-fibers | Radiology Reference Article | Radiopaedia.org". Radiopaedia. Retrieved 2020-11-10.
- ↑ Ali, Zarina S.; Van Der Voorn, J. Patrick; Powers, James M. (July 2007). "A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia--a role for oxidative damage". Journal of Neuropathology and Experimental Neurology. 66 (7): 660–672. doi:10.1097/nen.0b013e3180986247. ISSN 0022-3069. PMID 17620991.
- 1 2 3 4 5 6 Wider, C; Van Gerpen, J A.; DeArmond, S; Shuster, E A.; Dickson, D W.; Wszolek, Z K. (2009-06-02). "Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD)". Neurology. 72 (22): 1953–1959. doi:10.1212/WNL.0b013e3181a826c0. ISSN 0028-3878. PMC 2843560. PMID 19487654.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Konno, T.; Yoshida, K.; Mizuta, I.; Mizuno, T.; Kawarai, T.; Tada, M.; Nozaki, H.; Ikeda, S.-I.; Onodera, O.; Wszolek, Z. K.; Ikeuchi, T. (January 2018). "Diagnostic criteria for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia due to CSF1R mutation". European Journal of Neurology. 25 (1): 142–147. doi:10.1111/ene.13464. PMC 5741468. PMID 28921817.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 Sundal, Christina; Wszolek, Zbigniew K. (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "CSF1R-Related Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 22934315, retrieved 2020-11-11
- ↑ Van Gerpen, J. A.; Wider, C.; Broderick, D. F.; Dickson, D. W.; Brown, L. A.; Wszolek, Z. K. (2008-09-15). "Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids". Neurology. 71 (12): 925–929. doi:10.1212/01.wnl.0000325916.30701.21. ISSN 0028-3878. PMC 2843529. PMID 18794495.
- ↑ Sundal, C.; Van Gerpen, J. A.; Nicholson, A. M.; Wider, C.; Shuster, E. A.; Aasly, J.; Spina, S.; Ghetti, B.; Roeber, S.; Garbern, J.; Borjesson-Hanson, A. (2012-08-07). "MRI characteristics and scoring in HDLS due to CSF1R gene mutations". Neurology. 79 (6): 566–574. doi:10.1212/wnl.0b013e318263575a. ISSN 0028-3878. PMC 3413763. PMID 22843259.
- ↑ Konno, T.; Yoshida, K.; Mizuno, T.; Kawarai, T.; Tada, M.; Nozaki, H.; Ikeda, S.-I.; Nishizawa, M.; Onodera, O.; Wszolek, Z. K.; Ikeuchi, T. (2017). "Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation". European Journal of Neurology. 24 (1): 37–45. doi:10.1111/ene.13125. ISSN 1468-1331. PMC 5215554. PMID 27680516.
- ↑ Wong, Janice C.; Chow, Tiffany W.; Hazrati, Lili-Naz (2011). "Adult-Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia Can Present as Frontotemporal Dementia Syndrome". Dementia and Geriatric Cognitive Disorders. 32 (2): 150–158. doi:10.1159/000331422. ISSN 1420-8008. PMID 21986056.
- 1 2 3 4 5 "UpToDate". www.uptodate.com. Retrieved 2020-11-11.
- 1 2 3 4 Eichler, Florian S.; Li, Jiankang; Guo, Yiran; Caruso, Paul A.; Bjonnes, Andrew C.; Pan, Jessica; Booker, Jessica K.; Lane, Jacqueline M.; Tare, Archana; Vlasac, Irma; Hakonarson, Hakon (June 2016). "CSF1R mosaicism in a family with hereditary diffuse leukoencephalopathy with spheroids". Brain. 139 (6): 1666–1672. doi:10.1093/brain/aww066. ISSN 0006-8950. PMC 4892751. PMID 27190017.