Adenosine monophosphate deaminase deficiency type 1
| Adenosine monophosphate deaminase deficiency type 1 | |
|---|---|
| Other names: Myoadenylate deaminase deficiency | |
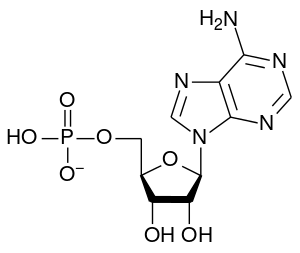 | |
| Adenosine monophosphate | |
Adenosine monophosphate deaminase deficiency type 1, (or Myoadenylate deaminase deficiency[1][2])is a human metabolic disorder in which the body consistently lacks the enzyme AMP deaminase,[3] in sufficient quantities. This may result in exercise intolerance, muscle pain and muscle cramping.
In virtually all cases, the deficiency has been caused by an SNP mutation, known as rs17602729 or C34T. While it was initially regarded as a recessive (or purely homozygous) disorder, some researchers have reported the existence of similarly deleterious effects from the heterozygous form of the SNP.[4] In the homozygous form of the mutation, a single genetic base (character) has been changed from cytosine ("C") to thymine ("T") on both strands of Chromosome 1 – in other words, "C;C" has been replaced by "T;T". A rarer but analogous condition, in which two guanine bases ("G;G") bases (in the unmutated form) have been changed to adenine ("A;A") has also been identified. While there has been no consensus on the effects of the heterozygous form – either "C;T" or "A;G" – some evidence has been found that it too has caused AMPD1 deficiency.[4] In addition, some sources have suggested the existence of a rare, acquired form of AMPD1 deficiency.
AMPD1 deficiency is caused by a defect in the mechanism for production of AMP deaminase – an enzyme that converts adenosine monophosphate (AMP) to inosine monophosphate (IMP).[5] While the deficiency affects approximately 1–2% of people in populations of predominantly European descent,[5] the disorder appears to be considerably rarer in Asian populations.[5]
Symptoms and signs
For reasons that are not understood, many people with defective variants of the AMPD genes are asymptomatic, while others have symptoms including exercise intolerance, and/or muscle pain and cramping.[3]
- Fatigue
- MADD lowers aerobic power output, so increased anaerobic power is needed to perform the same amount of work.
- Without myoadenylate deaminase, heavy activity causes adenosine to be released into the cell or perfused into the surrounding tissues. Fatigue and sedation after heavy exertion can be caused by excess adenosine in the cells which signals muscle fiber to feel fatigued. In the brain, excess adenosine decreases alertness and causes sleepiness. In this way, adenosine may play a role in fatigue from MADD.[6]
- Recovery from over-exertion can be hours, days or even months. In cases of rhabdomyolysis, which is the rapid breakdown of muscle fibers, time to recovery is dependent on duration and intensity of original activity plus any excess activity during the recovery period.
- Muscle pain
- Muscle pain from MADD is not well understood, but is partially due to high levels of lactate. Increased levels of free adenosine temporarily decrease pain, allowing over-exertion without awareness.[7] The over exertion can cause mild to severe cases of rhabdomyolysis, which is painful.[8]
- Adenosine mediates pain through adenosine receptors. MADD causes an increase of free adenosine during heavy activity which may cause exercise-induced muscle pain. Over time, excess free adenosine down-regulates primary A1 adenosine receptors, leading to increased muscle pain. Secondary receptors (A3) increase peripheral inflammation, which also increases pain.[9][10]
- Muscle cramping
- The cause of cramping is unknown, but may be related to elevated lactate, increased calcium signaling across the sarcoplasmic reticulum caused by membrane instability from reduced levels of ATP, or increased levels of free adenosine.[11]
- Muscle weakness
- Muscle weakness is not a major symptom, though the progressive effects of chronic muscle damage from rhabdomyolysis will eventually cause significant weakness. Similarly, the long-term metabolic effects may result in nerve damage.[8]
Potential complications
There is an increased risk that statin (cholesterol-reducing drugs) will cause myopathy (muscle weakness) in individuals with MADD.[12]
Anesthesia has the potential to cause malignant hyperthermia, an uncontrolled increase in body temperature, and permanent muscle damage in patients with MADD. Individuals with MADD are advised to notify their anesthesiologist about their condition prior to surgery.[8]
In most cases where myopathy is present with MADD, a second muscle disease is present and symptoms are worse than either disease in isolation.[13][14]
Causes
AMP deaminase is an enzyme that converts adenosine monophosphate (AMP) to inosine monophosphate (IMP), freeing an ammonia molecule in the process. It is a part of the metabolic process that converts sugar, fat, and protein into cellular energy. In order to use energy, a cell converts one of the above fuels into adenosine triphosphate (ATP) via the mitochondria. Cellular processes, especially muscles, then convert the ATP into adenosine diphosphate (ADP), freeing the energy to do work.
During heavy or prolonged mild to moderate activity, other enzymes convert two molecules of ADP into one ATP molecule and one AMP molecule, making more ATP available to supply energy. AMP is normally converted into IMP by myoadenylate deaminase—so myoadenylate deaminase deficiency reduces energy that would be available to the cell through the purine nucleotide cycle. Instead of being converted to IMP, the AMP builds up in the cells of affected individuals, spills into the blood, and is eventually metabolized in the liver. In persons with a defective enzyme, 5'-nucleotidase removes the ribose and phosphorus from AMP, increasing levels of adenosine measured in muscle cells by ~16–25×, after exercise.[15][16]
Mechanism
This failure to deaminate the AMP molecules has three major effects. First, significant amounts of AMP are lost from the cell and the body. Second, ammonia is not freed when the cell does work. Third, the level of IMP in the cell is not maintained.
- The first effect—the loss of AMP—is mostly significant because AMP contains ribose, a sugar molecule that is also used to make DNA, RNA, and some enzymes. Though the body can manufacture some ribose and obtain more from RNA-rich sources such as beans and red meat, this loss of ribose due to MADD is sometimes sufficient to create a shortage in the body, resulting in symptoms of severe fatigue and muscle pain. This outcome is especially likely if the individual regularly exercises vigorously or works physically over a period of weeks or months.
- The second effect, the absence of ammonia, is not well understood. It may result in a reduction of the amount of fumarate available to the citric acid cycle, and it may result in lower levels of nitric oxide (a vasodilator) in the body, reducing blood flow and oxygen intake during vigorous exercise, though this may be offset by increased levels of adenosine, another vasodilator.[17]
- The third effect, the reduction in IMP, is also not well understood. It may somehow result in a reduction in the amount of lactic acid produced by the muscles, though serum lactate is typically slightly elevated with MADD.
The following is a very simplified model of what may be going on inside a muscle cell with AMPD deficiency. There are two major semi-stable states: one with intra-cellular glycogen available, and one with glycogen exhausted. Both states are modified by how much the citric acid cycle is down-regulated by default.
Start from the state where glycogen is available and the citric acid cycle is severely down-regulated. Once the cell has received a non-trivial load, and has expended the phosphocreatine reserve, a small quantity of ATP will become discharged down to AMP. AMP will instantly up-regulate myophosphorylase, which will start liberating glucose from glycogen and make it available to the glycolytic pathway, producing pyruvate and recharging AMP back to ATP. Due to the greater availability of pyruvate as a substrate, and pyruvate also contributing a citric acid cycle intermediate, α-ketoglutarate, while consuming glutamate, the citric acid cycle will also speed up. The combination of glycolysis and the citric acid cycle now balances ATP production with ATP demand and the pool of AMP does not grow further. Because all pyruvate is not burned down in the citric acid cycle—a consequence of the pyruvate's concentration regulating its burning at this moment—the excess is converted to lactate and passed into blood as lactic acid.
In muscle cells with normal AMPD activity, the purine nucleotide cycle would now start to gradually add fumarate to the pool of the citric acid cycle intermediates. This would decrease the excess rate of pyruvate production by increasing its consumption, increase the rate of AMP recharge to ATP by the citric acid cycle, and consequently reduce liberation of glucose from glycogen, until increased supply of blood-borne fuels allows to shut down glycogenolysis completely.
In muscle cells with AMPD deficiency, ATP production rate of the citric acid cycle will not be synchronized with ATP demand. It was demonstrated,[18] that muscle cells, that lack AMPD1, stock and consume significantly more glutamate, and produce more alanine in this state, compared to healthy controls, which indicates occurrence of a higher concentration of pyruvate in the cell during exercise. The pool of AMP also grows bigger than in the controls, which would cause higher rate of glucose liberation from glycogen.
This state can last for as long as glycogen is available, and can be prolonged by constantly eating carbohydrate-rich food. If the load on muscles is greater than the body's ability to recycle lactate back into glucose, lactate will start to build up in the blood. Once lactate reaches its renal re-absorption threshold (5–6 mmol/L in general population), it gets lost to urine, wasting many calories (and producing bright matte yellow particles on surfaces where urine dries). At about the same time the kidney will also start correcting blood acidity by acidifying urine. Overly acidic urine causes irritation that feels like a frequent urge to urinate (with little volume) and a "hot" urine.
In order to excrete lactate, kidney must also excrete magnesium as an obligatory cation, which may lead to acute and chronic magnesium deficiency. Supplementary magnesium in the form of lactate or citrate may be rapidly lost in the same way. Because magnesium is essential to aerobic metabolism, over time, magnesium loss may lead to a vicious cycle, where the citric acid cycle is further down-regulated, lactate production is increased, and magnesium loss is increased again.
Although probably unrelated to AMPD deficiency, if the person happens to have a high load of d-lactate in the blood (mostly from food and colonic fermentation), the precipitate, the lactate loss and the magnesium loss may occur even before l-lactate (mostly from muscles) reaches its renal re-absorption threshold. This happens because l-lactate and d-lactate compete with each other for renal re-absorption, and because d-lactate has a significantly lower renal re-absorption threshold, <1 mmol/L.
In order to keep the excreted metabolites in solution, the kidney also has to excrete water. This is in contrast to complete oxidation of lactic acid, which would actually yield metabolic water for the body. This may lead to onset of acute thirst some tens of minutes into exercise in this state, if the water balance in the body was neutral initially.
If the muscle load is small, lactate is mostly recycled back into glucose or burned by other cells in the body. However the newly generated glucose is made available to all cells in the body, not just to muscle cells. The ability of the body to assimilate lactate may also be diminished, if working muscle cells cannot take up glucose from blood, due to myophosphorylase maintaining a higher concentration of it inside loaded cells, and if liver has already filled its glycogen stores up to capacity. So, ultimately, in this state, working muscle cells are destined to lose all glycogen. AMP breakdown to adenosine in this state is minor, because the pool of AMP is kept small by the vigorous regulatory action of myophosphorylase. Maximum continuous exertion is limited by the onset of burning sensation from lactate accumulation in muscles.
Eventually, all glycogen is exhausted, and the muscle cell enters another semi-stable state. During this transition, up-regulation of the citric acid cycle due to abundance of pyruvate is reversed, and a substantial part of the ATP pool is necessarily discharged down to AMP,[19] which allows the citric acid cycle to be sped up by some other mechanism (perhaps by the allosteric mechanism that reacts to the lower concentration of ATP, or by amplification of the residual AMPD activity by the bloated AMP pool), until ATP production is balanced with ATP consumption. AMP conversion to adenosine, excretion to the blood (as AMP and its various metabolites), further conversion to uric acid and excretion to urine becomes significant for some time, until all AMP is eliminated from the muscle cell. The muscle movements become noticeably less precise. Breathing slows down, and from this moment reacts very weakly to the load and not at all to the perceived effort. It becomes hard to rapidly increase the load on a muscle, as in McArdle's disease, and such a rapid load increase will dump even more purines into blood and urine (looking like translucent or rust-colored sharp shiny crystals and being highly irritating). The same situation would occur if blood flow to muscle cells becomes insufficient (except that somewhat less AMP is spilled over, and somewhat more of it is metabolized inside the muscle cell). On the other hand, there will be no lasting muscle pain from lactate, and continuous aerobic activity is possible. Oxidation of odd-numbered saturated fatty acids may provide another mechanism, although very gradual, to up-regulate citric acid cycle during exercise.
As the short-term regulation of ATP production becomes very weak after the exhaustion of glycogen, the medium-term regulation becomes enabled, but with a progressively weaker authority at higher purine nucleotide energy charge levels, which causes some differences in symptoms compared to McArdle's. In McArdle's, the highly active AMP deaminase, which additionally experiences amplification from the bloated AMP pool due to the lack of the moderating effect of myophosphorylase, is able to produce a readily observable "second wind" phenomenon almost exactly 7 minutes after a significant load increment. In AMPD deficiency, glycogen-less muscles will feel mostly the same by the time they become able to take another load increment. A load decrement may produce some sense of relief, though, if the pool of the citric acid cycle intermediates built up so far is sufficient to maintain full purine nucleotide energy charge at the lower load.
It is unclear, what, if anything, does it take to unknowingly trigger rhabdomyolysis at this point, assuming the muscle cell is otherwise healthy. Adenosine production and lack of ammonia overproduction seem to strongly suppress rhabdomyolysis down to the purine nucleotide energy charge level, where the cell is able to signal pain, or where individual muscle fibers start cramping (fail to relax from contraction in sync with the rest of the muscle), or the whole muscle fails to contract (when walking quickly downhill), allowing the person to appropriately modulate the exertion.
Most of the AMP probably spills into blood unchanged, and is gradually returned to the muscle cell, if its concentration there falls due to gradual recharge to ATP. The blood thus plays a role of a big AMP buffer. Idle muscles may also take up some free AMP. The spillover also limits, how much the residual AMPD activity can be amplified in this state. Thus, it may take the residual AMPD activity less time to build up citric acid cycle intermediates, when the whole body is warmed up for an exercise at the same time, rather than a specific group of muscles needed in the exercise.
In case of leg muscles, where circulation is substantially dependent on their cyclical contraction when the body is upright, a small but useful degree of initial up-regulation of the citric acid cycle may be achieved just by standing still for a few minutes. It is most useful when a long period of rest, or sitting in a vehicle, must be followed by brisk walking.
If the person keeps standing still for longer, rather than restoring circulation in the leg muscles (e.g. by sitting down, walking, or cycling), the ammonia, produced by the amplified residual AMPD activity, may accumulate in the muscle cells and in the surrounding tissues to toxic levels, and may also indirectly affect other organs. There is little or no warning for nearing toxicity, because the purine nucleotide energy charge is still relatively high, the leg muscles do not cramp, and remain functional. In contrast, while muscle glycogen is available, accumulation of lactic acid in this situation would produce a noticeable sensation. On the other hand, in persons with balanced AMPD and myophosphorylase activities in muscle cells, lactic acid and ammonia are produced simultaneously, counteracting each other's effects to some degree.
Some seldom used but strong voluntary muscles, such as those involved in "pushing" during the act of defecation, are not tuned for aerobic mode, and may dump plenty of purines during their short work routine, if it happens in this state.
If a food containing even small but perceivable amount of sugar (simple sugars or disaccharides that can be tasted sweet, or starch that is at least minimally hydrolyzed by salivary amylase, or even some non-sugar sweeteners) is eaten in this state, there may be a period of time after it enters stomach and before bulk absorption occurs, when continuous exercise becomes very hard, and easily triggers rhabdomyolysis. It probably happens because the digestive system senses and signals forthcoming delivery of sugars, inhibiting fatty acid release and oxidation, and starving glycogen-less muscle cells of the sole available source of energy. Even simple continuous exercise, like walking or washing dishes by hand right after the meal, may trigger rhabdomyolysis in the exercising muscles. This rhabdomyolysis is probably not of exertional, but of hypoglycemic nature, as loaded glycogen-less muscles can rapidly remove glucose from blood, and the normal mechanism of glucose homeostasis lacks the required responsiveness or capacity to prevent hypoglycemia. The broken down myocytes probably do not yield much glucose. Unlike the case with exertional rhabdomyolysis, there is no warning. At rest, however, the liver will effortlessly cover the whole body energy needs until absorption of carbohydrates occurs.
If a large group of muscles is still actively drawing fuels from blood after the most recent continuous exercise, in order to replenish the ATP and the phosphocreatine reserve, it may become sour without any additional exercise by the time the carbohydrate meal is finished.
If the carbohydrate meal consists of a food (which doesn't need to be carbohydrate itself), that requires prolonged vigorous chewing, and then some time to be digested, for instance parboiled long grain rice, chewing may suddenly become very slow and difficult halfway through the meal.
When a carbohydrate-rich food has been eaten before AMP has been eliminated from muscle cells, when bulk absorption starts, plenty of glucose becomes available in the blood, is taken up by muscle cells, is added to the glycogen store, but then immediately becomes liberated by the still up-regulated myophosphorylase. The resulting excess of glucose is metabolized down to lactic acid (the body cannot increase aerobic metabolism in an instant), recharging all AMP to ATP. The lactate is dumped back into blood and urine. The higher the glycemic index of the food is, the greater proportion of carbohydrates (and calories) is wasted into urine. If the person is at rest at this moment, and pays attention, a sudden increase in the breathing frequency due to lactate dumping is readily observable. If the rise in the blood lactate is particularly sharp, and the person happened to be breathing slowly, a heart palpitation may sometimes be observable. The lactic acidosis with palpitation may also occur during sleep, if stomach emptying has been delayed e.g. due to digestion requirements of the food or its high volume, and the person went to sleep before absorption has started. In this case the person will be awaken in a state of distress, with rapid breathing. The person may recall seeing a nightmare. Delayed stomach emptying creates especially favorable conditions for the shock lactic acidosis, because the digestive system may meanwhile still inhibit fatty acid release and oxidation, helping more muscles to run out of glycogen in those persons, who are otherwise still able to maintain its stores between meals. It has been experimentally demonstrated,[20] that delayed gastric emptying prolongs the duration of the GLP-1 signal.
Notably, a small quantity of dietary fructose does not produce this effect (the lactic acidosis), as it is captured by liver and may be fully expended for replenishing liver glycogen.
Once all AMP has been recharged to ATP, and glycogen stores allowed to replenish, the cell transitions back to the unmodified original state.
If carbohydrate-rich food is not consumed in this state, AMP elimination from the cell eventually completes, glycogen stores can be replenished again, and the cell transitions back to the original state but with reduced ATP pool and an up-regulated citric acid cycle.
It may be especially important to have adequate dietary iodine in the glycogen-less state, so that stomach emptying is not excessively delayed, the up-regulation of the citric acid cycle in muscle cells in response to a load increment is not too slow, and the muscles can each time accept a bigger load increment relative to the perceived effort.
Diagnosis
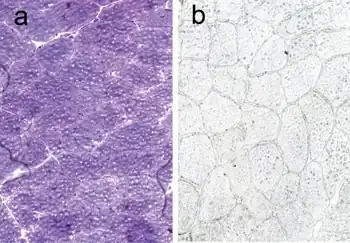
The diagnosis of MADD needs to be considered in patients who have exercise induced myalgia, cramps and sometimes weakness. A mildly elevated creatine kinase may also occur. Exclusion of other muscular diseases such as McArdle's Disease and carnitine cycle abnormalities should occur. MADD may be identified if there is a lack of ammonia rise after forearm exercise testing. The diagnosis may then be confirmed with genetic testing.
Treatment
It is important for MADD patients to maintain strength and fitness without exercising or working to exhaustion. Learning this balance may be more difficult than normally, as muscle pain and fatigue may be perceived differently from normal individuals.[21]
Symptomatic relief from the effects of MADD may sometimes be achieved by administering ribose orally at a dose of approximately 10 grams per 100 pounds (0.2 g/kg) of body weight per day, and exercise modulation as appropriate. Taken hourly, ribose provides a direct but limited source of energy for the cells. Patients with myoadenylate deaminase deficiency do not retain ribose during heavy exercise, so supplementation may be required to rebuild levels of ATP.[22][23]
Creatine monohydrate could also be helpful for AMPD patients, as it provides an alternative source of energy for anaerobic muscle tissue and was found to be helpful in the treatment of other, unrelated muscular myopathies.[24]
See also
- Inborn Errors of Metabolism
- Metabolic Myopathies
- Purine Nucleotide Cycle
References
- ↑ "Adenosine monophosphate deaminase deficiency (Concept Id: C2931781) - MedGen - NCBI". www.ncbi.nlm.nih.gov. Archived from the original on 19 September 2023. Retrieved 16 September 2023.
- ↑ "Adenosine monophosphate deaminase deficiency". orphanet. Archived from the original on 23 March 2023. Retrieved 16 September 2023.
- 1 2 "Metabolic Diseases of Muscle - Myoadenylate deaminase deficiency | Muscular Dystrophy Association". Muscular Dystrophy Association. 18 December 2015. Archived from the original on 11 July 2017. Retrieved 10 June 2017.
- 1 2 "SNPedia, 2019, rs17602729 (23 October 2021)". Archived from the original on 25 March 2023. Retrieved 15 August 2023.
- 1 2 3 "Adenosine monophosphate deaminase deficiency". Genetics Home Reference. United States National Library of Medicine. July 2008. Archived from the original on 2020-09-27. Retrieved 2023-08-15.
- ↑ Morisaki, H; Morisaki, T (2008). "AMPD genes and urate metabolism". Nihon Rinsho. Japanese Journal of Clinical Medicine. 66 (4): 771–7. PMID 18409530.
- ↑ Belfrage, Måns; Sollevi, Alf; Segerdahl, Märta; Sjölund, Karl-Fredrik; Hansson, Per (1995). "Systemic Adenosine Infusion Alleviates Spontaneous and Stimulus Evoked Pain in Patients with Peripheral Neuropathic Pain". Anesthesia & Analgesia. 81 (4): 713–7. doi:10.1097/00000539-199510000-00010. PMID 7573999. S2CID 23963005. Archived from the original on 2023-09-19. Retrieved 2023-08-15.
- 1 2 3 "Facts About Metabolic Diseases of Muscle" (PDF). Muscular Dystrophy Association. December 2009. Archived from the original (PDF) on 2011-09-27.
- ↑ Li, Xinhui; Bantel, Carsten; Conklin, Dawn; Childers, Steven R.; Eisenach, James C. (2004). "Repeated Dosing with Oral Allosteric Modulator of Adenosine A1 Receptor Produces Tolerance in Rats with Neuropathic Pain". Anesthesiology. 100 (4): 956–61. doi:10.1097/00000542-200404000-00028. PMID 15087633. S2CID 35764254.
- ↑ Fredholm, B. B.; Halldner, L.; Johansson, C.; Schulte, G.; Lövdahl, C.; Thorén, P.; Dunwiddie, T. V.; Masino, S. A.; Poelchen, W.; Diao, L.; Illes, P.; Zahniser, N. R.; Valen, G.; Tokuno, S.; Sommerschild, H.; Giménez-Llort, L.; Fernández-Teruel, A.; Escorihuela, R. M.; Wiesenfeld-Hallin, Z.; Xu, X. J.; Hårdemark, A.; Herlenius, E.; Pekny, M.; Gebré-Medhin, S.; Brown, R.; Ollerstam, A.; Persson, A. E. G.; Skøtt, O.; Johansson, B. R. (2003). "Consequences of eliminating adenosine A1 receptors in mice". Drug Development Research. 58 (4): 350–353. doi:10.1002/ddr.10170. S2CID 84898432.
- ↑ Blazev R, Lamb GD (December 1999). "Adenosine inhibits depolarization-induced Ca(2+) release in mammalian skeletal muscle". Muscle Nerve. 22 (12): 1674–83. doi:10.1002/(SICI)1097-4598(199912)22:12<1674::AID-MUS9>3.0.CO;2-0. PMID 10567080. S2CID 41846255.
- ↑ Vladutiu, Georgirene D.; Simmons, Zachary; Isackson, Paul J.; Tarnopolsky, Mark; Peltier, Wendy L.; Barboi, Alexandru C.; Sripathi, Naganand; Wortmann, Robert L.; Phillips, Paul S. (2006). "Genetic risk factors associated with lipid-lowering drug-induced myopathies". Muscle & Nerve. 34 (2): 153–62. doi:10.1002/mus.20567. PMID 16671104. S2CID 22609911.
- ↑ Vockley, Jerry; Rinaldo, Piero; Bennett, Michael J.; Matern, Dietrich; Vladutiu, Georgirene D. (2000). "Synergistic Heterozygosity: Disease Resulting from Multiple Partial Defects in One or More Metabolic Pathways". Molecular Genetics and Metabolism. 71 (1–2): 10–8. doi:10.1006/mgme.2000.3066. PMID 11001791.
- ↑ Sabina, Richard L. (2000). "MYOADENYLATE DEAMINASE DEFICIENCY: A Common Inherited Defect with Heterogeneous Clinical Presentation". Neurologic Clinics. 18 (1): 185–94. doi:10.1016/S0733-8619(05)70184-5. PMID 10658174.
- ↑ Sabina, R L; Swain, J L; Olanow, C W; Bradley, W G; Fishbein, W N; Dimauro, S; Holmes, E W (1984). "Myoadenylate deaminase deficiency. Functional and metabolic abnormalities associated with disruption of the purine nucleotide cycle". Journal of Clinical Investigation. 73 (3): 720–30. doi:10.1172/JCI111265. PMC 425074. PMID 6707201.
- ↑ Loh, Evan (2000). "AMPD1 Genotype Predicts Survival in Patients with Heart Failure". Japanese Circulation Society. Archived from the original on 2021-05-18. Retrieved 2023-08-15.
- ↑ Costa F, Biaggioni I (May 1998). "Role of nitric oxide in adenosine-induced vasodilation in humans". Hypertension. 31 (5): 1061–4. doi:10.1161/01.HYP.31.5.1061. PMID 9576114.
- ↑ Tarnopolsky, Mark A.; Parise, Gianni; Gibala, Martin J.; Graham, Terry E.; Rush, James W. E. (15 June 2001). "Myoadenylate deaminase deficiency does not affect muscle anaplerosis during exhaustive exercise in humans". The Journal of Physiology. 533 (3): 881–889. doi:10.1111/j.1469-7793.2001.t01-1-00881.x. PMC 2278656. PMID 11410643.
- ↑ Sabina, Richard L.; Swain, Judith L.; Patten, Bernard M.; Ashizawa, Tetsuo; O'Brien, William E.; Holmes, Edward W. (1 December 1980). "Disruption of the Purine Nucleotide Cycle". Journal of Clinical Investigation. 66 (6): 1419–1423. doi:10.1172/JCI109995. PMC 371628. PMID 7440723.
- ↑ Frost, GS; Brynes, AE; Dhillo, WS; Bloom, SR; McBurney, MI (February 2003). "The effects of fiber enrichment of pasta and fat content on gastric emptying, GLP-1, glucose, and insulin responses to a meal". European Journal of Clinical Nutrition. 57 (2): 293–8. doi:10.1038/sj.ejcn.1601520. PMID 12571662.
- ↑ Lang, Robert (1998). "What is this Adenosine stuff?". Australasian Anaesthesia. ISSN 1032-2515. Archived from the original on 2011-10-20. Retrieved 2011-08-14.
- ↑ Wagner, D.R.; Gresser, U.; Zöllner, N. (1991). "Effects of Oral Ribose on Muscle Metabolism during Bicycle Ergometer in AMPD-Deficient Patients". Annals of Nutrition and Metabolism. 35 (5): 297–302. doi:10.1159/000177660. PMID 1776826.
- ↑ Zöllner, N.; Reiter, S.; Gross, M.; Pongratz, D.; Reimers, C. D.; Gerbitz, K.; Paetzke, I.; Deufel, T.; Hübner, G. (1986). "Myoadenylate deaminase deficiency: Successful symptomatic therapy by high dose oral administration of ribose". Klinische Wochenschrift. 64 (24): 1281–90. doi:10.1007/BF01785710. PMID 3102830. S2CID 35397362.
- ↑ Tarnopolsky, Mark A. (2007). "Clinical Use of Creatine in Neuromuscular and Neurometabolic Disorders". In Salomons, Gajja S.; Wyss, Markus (eds.). Creatine and Creatine Kinase in Health and Disease. Subcellular Biochemistry. Vol. 46. pp. 183–204. doi:10.1007/978-1-4020-6486-9_10. ISBN 978-1-4020-6485-2. PMID 18652078. Archived from the original on 2023-09-19. Retrieved 2023-08-15.
Further reading
- Fischer, H.; Esbjornsson, M.; Sabina, R. L.; Stromberg, A.; Peyrard-Janvid, M.; Norman, B. (2007). "AMP deaminase deficiency is associated with lower sprint cycling performance in healthy subjects". Journal of Applied Physiology. 103 (1): 315–22. doi:10.1152/japplphysiol.00185.2007. PMID 17463303.
- Skalova, K; Luptak, I; Turcani, M; Hulin, I (2002). "Adenosine and cardioprotection: what can we learn from nature's genetic polymorphism?" (PDF). Bratislavske Lekarske Listy. 103 (6): 187–93. PMID 12448564. Archived from the original (PDF) on 2012-03-23. Retrieved 2011-08-12.
External links
| Classification | |
|---|---|
| External resources |
|