Nakajo syndrome
| Nakajo syndrome | |
|---|---|
| Other names: Nodular erythema with digital changes, Amyotrophy-fat tissue anomaly syndrome, Secondary hypertrophic osteoperiostosis with pernio | |
 | |
| Nakajo syndrome has an autosomal recessive pattern of inheritance. | |
| Specialty | Medical genetics |
Nakajo syndrome, also called nodular erythema with digital changes,[1] is a rare autosomal recessive congenital disorder first reported in 1939 by A. Nakajo in the offspring of consanguineous (blood relative) parents.[2][3] The syndrome can be characterized by erythema (reddened skin), loss of body fat in the upper part of the body, and disproportionately large eyes, ears, nose, lips, and fingers.[1]
Signs and symptoms
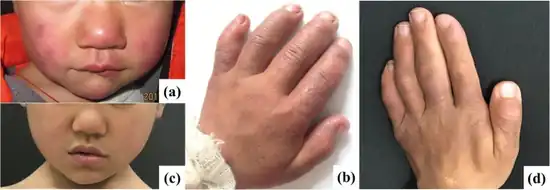
Signs of the disease begins during early childhood with individuals developing red, swollen lumps (nodular erythema) on the skin especially during colder weather, frequent fevers, and elongated fingers and toes with widened and rounded tips (clubbing).[4]
Later in childhood, individuals with the disease develop join pain, and joint deformities (contractures) that limit movement predominantly in the hands, wrists, and elbows. They additional have weakness and wasting of muscle and fat loss that worsens over time. This leads to an extremely thin (emaciated) appearance in the face, chest, and arms.[4]
Other symptoms of Nakajo-Nishimura syndrome are enlarged liver and spleen (hepatosplenomegaly), shortage of red blood cells (anemia), reduced levels of platelet blood cells (thrombocytopenia), and calcification in the basal ganglia area of the brain. There have been cases of intellectual disability in affected individuals.[4]
Nakajo-Nishimura syndrome has overlapping symptoms with joint contractures, muscular atrophy, microcytic anemia, and panniculitis-induced lipodystrophy (JMP) syndrome and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. These conditions are all caused by mutations in the PSMB8 and characterized by skin abnormalities and lipodystrophy. Although these conditions are considered different diseases, some researchers believe they represent different forms of a single condition.[4]
Genetics
Nakajo syndrome is inherited in an autosomal recessive manner.[3] This means the defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. This gene is called PSMB8 and is located on chromosome 6 at 6p21.32.[5] This gene codes for a subunit called immunoproteasomes that are found immune system cells. These cells are crucial for identifying viral proteins. It has been noted that malfunctions in PSMB8 cause the destruction of proteins which leads to muscle degradation and fat loss. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.[4] Although variations in severity has little clinical support, onset of Nakajo syndrome can vary from ages 1–18.[6]
Diagnosis
Typical diagnosis looks for at least 5 out of 8 proposed criteria for Nakajo Syndrome: an autosomal recessive inheritance pattern, pernio-like purplish lesions (on hands and feet), ''haunting'' nodular erythema, repetitive spiking fever, long clubbed fingers and toes with joint contractures, progressive upper body lipomuscular atrophy/emaciation, hepatosplenomegaly and basal ganglion calcification on computed tomography (CT) scans. These features are not always apparent until childhood. Histopathologic examination shows focal mononuclear cell infiltration with vasculopathy. Laboratory results include continually elevated C-reactive protein (CRP) levels and hyper-gamma-globulinemia. Autoantibody titers rise as the disease progresses in some but remain negative in others. Molecular genetic testing can detect a disease causing mutation, verifying diagnosis.[7]
Management
Currently, there are no treatments for Nakajo syndrome. However, available treatment options may help alleviate some of the symptoms. Existing steroids may help reduce inflammatory responses. Various immunosuppressive and anti-rheumatic have shown to have little or no temporal response in PRAAS patients. Ultimately, the progressive lipodystrophy caused by Nakajo syndrome continues despite treatment.[9]
Epidemiology
Two affected siblings reported by Tanaka were born from a consanguineous marriage.[10] Though a majority of cases originate in Japan, however two siblings reported by Garg were Portuguese and also born from a consanguineous marriage.[6]
References
- 1 2 Online Mendelian Inheritance in Man (OMIM): Nakajo syndrome - 256040
- ↑ Nakajo A (1939). "Secondary hypertrophic osteoperiostosis with pernio". J Derm Venerol. (in Japanese). 45: 77–86.
{{cite journal}}: CS1 maint: unrecognized language (link) - 1 2 Kitano Y, Matsunaga E, Morimoto T, Okada N, Sano S (August 1985). "A syndrome with nodular erythema, elongated and thickened fingers, and emaciation". Archives of Dermatology. 121 (8): 1053–1056. doi:10.1001/archderm.121.8.1053. PMID 4026345.
- 1 2 3 4 5 "Nakajo-Nishimura syndrome: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 2022-03-17. Retrieved 2022-03-17.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ↑ "PSMB8 proteasome 20S subunit beta 8 [ Homo sapiens (human) ]". NCBI Gene. National Library of Medicine (US), National Center for Biotechnology Information. Archived from the original on 2023-03-07. Retrieved 2023-10-27.
- 1 2 Garg A, Hernandez MD, Sousa AB, Subramanyam L, Martínez de Villarreal L, dos Santos HG, Barboza O (September 2010). "An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy". The Journal of Clinical Endocrinology and Metabolism. 95 (9): E58–E63. doi:10.1210/jc.2010-0488. PMC 2936059. PMID 20534754.
- ↑ "Nakajo Nishimura syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2022-03-21. Retrieved 2022-03-21.
- ↑ Ohmura, Koichiro (2019). "Nakajo-Nishimura syndrome and related proteasome-associated autoinflammatory syndromes". Journal of Inflammation Research. 12: 259–265. doi:10.2147/JIR.S194098. ISSN 1178-7031.
- ↑ McDermott A, Jacks J, Kessler M, Emanuel PD, Gao L (February 2015). "Proteasome-associated autoinflammatory syndromes: advances in pathogeneses, clinical presentations, diagnosis, and management". International Journal of Dermatology. 54 (2): 121–129. doi:10.1111/ijd.12695. PMID 25521013. S2CID 205189155.
- ↑ Tanaka M, Miyatani N, Yamada S, Miyashita K, Toyoshima I, Sakuma K, et al. (January 1993). "Hereditary lipo-muscular atrophy with joint contracture, skin eruptions and hyper-gamma-globulinemia: a new syndrome". Internal Medicine. 32 (1): 42–45. doi:10.2169/internalmedicine.32.42. PMID 8495043. S2CID 39807679.
External links
| Classification | |
|---|---|
| External resources |
|