Nuchal fibroma
| Nuchal fibroma | |
|---|---|
| Specialty | Oncology |
Nuchal-type fibroma is a rare benign proliferation involving the dermis and subcutaneous tissues, that is a collection of dense, hypocellular bundles of collagen with entrapped adipocytes and increased numbers of small nerves. It is no longer called a nuchal fibroma, but instead a "nuchal-type fibroma" since it develops in other anatomic sites. There is no known etiology.[1][2] The World Health Organization, 2020, classified nuchal fibromas as a specific tumor form in the category of benign fibroblastic and myofibroblastic tumors.[3]
Signs and symptoms
These lesions are generally asymptomatic, although patients give a long history of a solitary, superficial mass. The mass is usually in the neck (hence the name "nuchal-type"), but it can be seen in the extremities, lumbosacral area, buttocks, and face.[2][4] There is a strong association with diabetes mellitus and Gardner syndrome; in fact, it may be the initial manifestation of Gardner syndrome.[5]
Pathology

The tumors are unencapsulated and poorly circumscribed, showing a firm, white cut surface. Most tumors are about 3.5 cm, but can be up to 8 cm.[1] By microscopic examination, there are haphazardly arranged thick collagen fibers, with a low cellularity and no pleomorphism. There are usually entrapped fat cells, skeletal muscle, and peripheral nerves. The may be perineural fibrosis. The elastic fibers may be altered, which is why an elastofibroma is considered in the differential diagnosis.[1]
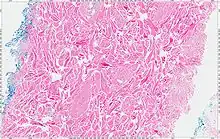
Immunohistochemistry
The tumor cells are strongly positive for vimentin, CD34, and sometimes with CD99. There is often (up to 2/3rds) a nuclear reaction with β-catenin.[1]
Diagnosis
Differential diagnoses
The differential diagnosis includes elastofibroma, fibrolipoma, desmoid-type fibromatoses, and nuchal fibrocartilaginous pseudotumor.[1]
Management
Simple excision is curative. However, in patients with Gardner syndrome, up to 45% will develop desmoid-type fibromatosis at other sites, and so this should be searched for and excluded. Patients can develop a recurrence, so follow-up is required.[2]
Epidemiology
This is a rare tumor, presenting over a wide age range, but usually in the 3rd to 5th decades of life. There is a slight male predilection, although this is not seen in syndrome associated patients. The most common site is the posterior neck, but may also be seen in other sites (extremities, lumbosacral area, buttocks, face).[1]
See also
References
- 1 2 3 4 5 6 Michal, M.; Fetsch, J. F.; Hes, O.; Miettinen, M. (1999). "Nuchal-type fibroma". Cancer. 85 (1): 156–163. doi:10.1002/(SICI)1097-0142(19990101)85:1<156::AID-CNCR22>3.0.CO;2-O. PMID 9921988.
- 1 2 3 Samadi, D. S.; McLaughlin, R. B.; Loevner, L. A.; Livolsi, V. A.; Goldberg, A. N. (2000). "Nuchal fibroma: A clinicopathological review". The Annals of Otology, Rhinology, and Laryngology. 109 (1): 52–55. doi:10.1177/000348940010900110. PMID 10651413.
- ↑ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
- ↑ Balachandran, K.; Allen, P. W.; MacCormac, L. B. (1995). "Nuchal fibroma. A clinicopathological study of nine cases". The American Journal of Surgical Pathology. 19 (3): 313–317. doi:10.1097/00000478-199503000-00009. PMID 7872429.
- ↑ Dawes, L. C.; La Hei, E. R.; Tobias, V.; Kern, I.; Stening, W. (2000). "Nuchal fibroma should be recognized as a new extracolonic manifestation of Gardner-variant familial adenomatous polyposis". The Australian and New Zealand Journal of Surgery. 70 (11): 824–826. doi:10.1046/j.1440-1622.2000.01958.x. PMID 11147449.
Further reading
Lester D. R. Thompson; Bruce M. Wenig (2011). Diagnostic Pathology: Head and Neck: Published by Amirsys. Hagerstown, MD: Lippincott Williams & Wilkins. pp. 8:44–45. ISBN 978-1-931884-61-7.