Parkin (protein)


Parkin is a 465-amino acid residue E3 ubiquitin ligase, a protein that in humans and mice is encoded by the PARK2 gene.[5][6] Parkin plays a critical role in ubiquitination – the process whereby molecules are covalently labelled with ubiquitin (Ub) and directed towards degradation in proteasomes or lysosomes. Ubiquitination involves the sequential action of three enzymes. First, an E1 ubiquitin-activating enzyme binds to inactive Ub in eukaryotic cells via a thioester bond and mobilises it in an ATP-dependent process. Ub is then transferred to an E2 ubiquitin-conjugating enzyme before being conjugated to the target protein via an E3 ubiquitin ligase.[7] There exists a multitude of E3 ligases, which differ in structure and substrate specificity to allow selective targeting of proteins to intracellular degradation.
In particular, parkin recognises proteins on the outer membrane of mitochondria upon cellular insult and mediates the clearance of damaged mitochondria via autophagy and proteasomal mechanisms.[8] Parkin also enhances cell survival by suppressing both mitochondria-dependent and -independent apoptosis. Mutations are associated with mitochondrial dysfunction, leading to neuronal death in Parkinson’s disease[9] and aberrant metabolism in tumourigenesis.[10]
Structure
The precise function of parkin is unknown; however, the protein is a component of a multiprotein E3 ubiquitin ligase complex which in turn is part of the ubiquitin-proteasome system that mediates the targeting of proteins for degradation. Mutations in this gene are known to cause a familial form of Parkinson's disease known as autosomal recessive juvenile Parkinson's disease (AR-JP). Moreover, parkin is described to be necessary for mitophagy (autophagy of mitochondria).
However, how loss of function of the parkin protein leads to dopaminergic cell death in this disease is unclear. The prevailing hypothesis is that parkin helps degrade one or more proteins toxic to dopaminergic neurons. Putative substrates of parkin include synphilin-1, CDC-rel1, cyclin E, p38 tRNA synthase, Pael-R, synaptotagmin XI, sp22 and parkin itself (see also ubiquitin ligase). Additionally, parkin contains a C-terminal motif that binds PDZ domains. Parkin has been shown to associate in a PDZ dependent manner with the PDZ domain containing proteins CASK and PICK1.
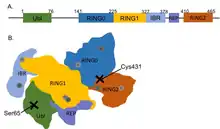
Like other members of the RING-between-RING (RBR) family of E3 ligases, parkin possesses two RING finger domains and an in-between-RING (IBR) region. RING1 forms the binding site for E2 Ub-conjugating enzyme while RING2 contains the catalytic cysteine residue (Cys431) that cleaves Ub off E2 and transiently binds it to E3 via a thioester bond.[8] Ub transfer is aided by neighbouring residues histidine His433, which accepts a proton from Cys431 to activate it, and glutamate Glu444, which is involved in autoubiquitination.[11] Together these form the catalytic triad, whose assembly is required for parkin activation.[12] Parkin also contains an N-terminal Ub-like domain (Ubl) for specific substrate recognition, a unique RING0 domain and a repressor (REP) region that tonically suppresses ligase activity.
Under resting conditions, the tightly coiled conformation of parkin renders it inactive, as access to the catalytic RING2 residue is sterically blocked by RING0, while the E2 binding domain on RING1 is occluded by Ubl and REP.[8] Activating stimuli disrupt these interdomain interactions and induce parkin to collapse along the RING1-RING0 interface.[12] The active site of RING2 is drawn towards E2-Ub bound to RING1, facilitating formation of the Ub-thioester intermediate. Parkin activation requires phosphorylation of serine Ser65 in Ubl by serine/threonine kinase, PINK1. Addition of a charged phosphate destabilises hydrophobic interactions between Ubl and neighbouring subregions, reducing autoinhibitory effects of this N-terminus domain.[13] Ser65Ala missense mutations were found to ablate Ub-parkin binding whilst inhibiting parkin recruitment to damaged mitochondria.[14] PINK1 also phosphorylates Ub at Ser65, accelerating its discharge from E2 and enhancing its affinity for parkin.[13]
Although structural changes following phosphorylation are uncertain, crystallisation of parkin revealed a cationic pocket in RING0 formed by lysine and arginine residues Lys161, Arg163 and Lys211 that forms a putative phosphate binding site.[15] Considering that RING0 is unique to parkin and that its hydrophobic interface with RING1 buries Cys431 in inactive parkin,[14] targeting of phosphorylated Ub and/or Ubl towards this binding niche might be critical in dismantling autoinhibitory complexes during parkin activation.
Function
Mitophagy
Parkin plays a crucial role in mitophagy and clearance of reactive oxygen species.[16] Mitophagy is the elimination of damaged mitochondria in autophagosomes, and is dependent on a positive feedback cycle involving synergistic action of parkin and PINK1. Following severe cellular insult, rundown of mitochondrial membrane potential prevents import of PINK1 into the mitochondrial matrix and causes it to aggregate on the outer mitochondrial membrane (OMM).[17] Parkin is recruited to mitochondria following depolarisation and phosphorylated by PINK1, which simultaneously phosphorylates Ub pre-conjugated to mitochondrial membrane proteins. PINK1 and Ub phosphorylation facilitate parkin activation and further assembly of mono- and poly-Ub chains.[13] Considering the proximity of these chains to PINK1, further phosphorylation of Ub at Ser65 is likely, potentiating parkin mobilisation and substrate ubiquitination in a self-reinforcing cycle.[8]
Parkin substrates include mitofusins Mfn1 and Mfn2, which are large GTPases that promote mitochondria fusion into dynamic, tubular complexes that maximise efficiency of oxidative phosphorylation.[18] However, upon mitochondrial damage, degradation of fusion proteins is necessary to separate them from the network via mitochondrial fission and prevent the corruption of healthy mitochondria.[19] Parkin is therefore required before mitophagy as it ubiquinates Mfn1/2, labelling it for proteasomal degradation. Proteomic studies identified additional OMM proteins as parkin substrates, including fission protein FIS, its adaptor TBC1D15 and translocase TOMM20 and TOMM70 that facilitate movement of proteins such as PINK1 across OMM.[20] Miro (or RHOT1/RHOT2) is an OMM protein critical for axonal transport, and may be ubiquitinated and targeted towards proteasomal degradation by parkin.[21] Miro breakdown produced a marked decrease in migration of compromised mitochondria along axons of mouse hippocampal neurons,[22] reinforcing the importance of parkin in segregating defective mitochondria from their functioning counterparts and limiting the spatial spread of mitochondrial dysfunction, prior to autophagy.
During mitophagy, parkin targets VDAC1, a voltage-gated anion channel that undergoes a conformational change upon mitochondrial membrane depolarisation, exposing a cytosolic domain for ubiquitination.[17] Silencing of VDAC1 expression in HeLa cells significantly reduced parkin recruitment to depolarised mitochondria and their subsequent clearance,[23] highlighting the critical role of VDAC1 as a selective marker of mitochondrial damage and instigator of mitophagy. Following Ub conjugation, parkin recruits autophagy receptors such as p62, TAX1BP1 and CALCOCO2, facilitating assembly of autophagosomes that digest defective mitochondria.[20]
Cell survival
Through activation of NF-κB signalling, parkin enhances survival and protects cells from stress-induced apoptosis. Upon cellular insult, parkin activates the catalytic HOIP subunit of another E3 ligase LUBAC. HOIP triggers assembly of linear Ub polymers on NF-κB essential modulator (NEMO), potentiating transcription of mitochondrial GTPase OPA1.[24] Increased OPA1 translation maintains cristae structure and reduces cytochrome C release from mitochondria, inhibiting caspase-mediated apoptosis. Importantly, parkin activates HOIP with greater potency than other LUBAC-associated factors HOIL-1 and sharpin,[25] meaning that parkin mobilisation significantly enhances tolerance to moderate stressors.
Parkin possesses DNA binding affinity and produces a dose-dependent reduction in transcription and activity of pro-apoptotic factor p53. Transfection of p53 promoter with truncated versions of parkin into SH-SY5Y neurons revealed that parkin directly binds to the p53 promoter via its RING1 domain.[26] Conversely, parkin may be a transcriptional target of p53 in H460 lung cells, where it mediates the tumour suppressor action of p53.[10] Considering its role in mitochondrial homeostasis, parkin aids p53 in maintaining mitochondrial respiration while limiting glucose uptake and lactate production, thus preventing onset of the Warburg effect during tumourigenesis.[27] Parkin further elevates cytosolic glutathione levels and protects against oxidative stress, characterising it as a critical tumour suppressor with anti-glycolytic and antioxidant capabilities.[10]
Clinical significance
Parkinson’s disease
PARK2 (OMIM *602544) is the parkin gene that may cause a form of autosomal recessive juvenile Parkinson disease (OMIM 600116) due to a mutation in the parkin protein. This form of genetic mutation may be one of the most common known genetic causes of early-onset Parkinson disease. In one study of patients with onset of Parkinson disease prior to age 40 (10% of all PD patients), 18% had parkin mutations, with 5% homozygous mutations.[28] Patients with an autosomal recessive family history of parkinsonism are much more likely to carry parkin mutations if age at onset is less than 20 (80% vs. 28% with onset over age 40).[29]
Patients with parkin mutations (PARK2) do not have Lewy bodies. Such patients develop a syndrome that closely resembles the sporadic form of PD; however, they tend to develop symptoms at a much younger age. In humans, loss-of-function mutations in parkin PARK2 gene have been implicated in 50% of inherited and 15% of juvenile-onset sporadic forms of Parkinson’s disease (PD).[16] While PD is traditionally regarded a late-onset neurodegenerative condition characterised by alpha-synuclein-enriched Lewy bodies, autosomal recessive PD due to parkin mutations is often early onset and lack the ubiquitinated protein deposits pathognomonic for sporadic PD.[21] Parkin-mutant PD could also involve loss of noradrenergic neurons in the locus coeruleus alongside the hallmark degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc).[30] However, its symptoms resembles those of idiopathic PD, with patients presenting with resting tremors, postural instability and bradykinesia.[9]
While mitochondria are essential for ATP generation in any eukaryotic cell, catecholaminergic neurons are particularly reliant on their proper function for clearance of reactive oxygen species produced by dopamine metabolism, and to supply high energy requirements of catecholamine synthesis.[17] Their susceptibility to oxidative damage and metabolic stress render catecholaminergic neurons vulnerable to neurotoxicity associated with aberrant regulation of mitochondrial activity, as is postulated to occur in both inherited and idiopathic PD. For example, enhanced oxidative stress in neurons, skeletal muscle and platelets, corresponding with reduced activity of complex I in the electron transport chain were reported in PD patients,[31] while deletions in the mitochondrial genome were found in the SNpc.[32]
In accordance with its critical role in mitochondrial quality control, more than 120 pathogenic, PD-inducing mutations have been characterised on parkin.[8] Such mutations may be hereditary or stochastic and are associated with structural instability, reduced catalytic efficiency and aberrant substrate binding and ubiquitination.[9] Mutations can generally be categorised into three groups, depending on their location. Firstly, those clustered around Zn-coordinating residues on RING and IBR might compromise structural integrity and impair catalysis.[12] A second class of mutations, including Thr240Arg, affect residues in and around the E2 binding site and alter autoinhibition of RING1 by REP.[33] Finally, Cys431Phe and Gly430Asp mutations impair ligase activity at the catalytic site and significantly reduce parkin function.[8]
The discovery of numerous non-mitochondrial parkin substrates reinforces the importance parkin in neuronal homeostasis, beyond its role in mitochondrial regulation. Potent neuroprotective abilities of parkin in attenuating dopaminergic neurotoxicity, mitochondrial swelling and excitotoxicity were demonstrated in cell cultures over-expressing parkin,[9] although the existence of such mechanisms at physiological parkin levels in vivo is yet unconfirmed. Another parkin substrate, synphilin-1 (encoded by SNCAIP), is an alpha-synuclein interacting protein that is enriched in the core of Lewy bodies and ubiquitinated by parkin in a manner abolished by familial PD-associated mutations.[34] Parkin might promote aggregation of alpha-synuclein and synphilin-1 into Lewy bodies, which are conjugated to Lys63-linked poly-Ub chains and directed towards autophagic degradation.[35] Parkin mutations therefore inhibit this mechanism, leading to toxic accumulation of soluble proteins that overloads the proteasome. Protein aggregation triggers neuronal toxicity, whilst accounting for lack of ubiquitinated Lewy bodies in parkin-mutant PD. Similarly, native parkin reduces death of SH-SY5Y neurons by ubiquitinating other Lewy body constituents, such as the p38 subunit of aminoacyl-tRNA synthetase complex[36] and far upstream element-binding protein 1[37] through addition of Lys48-linked poly-Ub chains and directing them towards proteasomal degradation. Parkin also influences axonal transport and vesicle fusion through ubiquitination of tubulin and synaptotagmin XI (SYT11) respectively, giving it a modulatory role in synapse function.[9]
Finally, parkin protects dopaminergic neurons from cytotoxicity induced by PD-mimetic 6-OHDA, mediated by suppression of neuronal p53 expression and its downstream activation of the apoptotic cascade.[26] Several PD-associated parkin mutations are localised to RING1 and might impair its ability to bind and downregulate the p53 promoter, leading to enhanced p53 expression.[38] Parkin-mutant PD patients also exhibit a four-fold elevation in p53 immunoreactivity,[26] insinuating that failure of parkin-mediated anti-apoptosis might be involved in etiology of PD.
Tumourigenesis
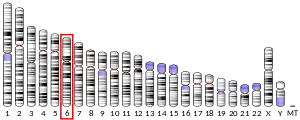
Consistent with parkin’s potent anti-tumourigenic abilities, negative mutations and deletions have been reported in various tumours. For example, PARK2 copy number was reduced in 85% of glioblastoma samples while lung cancers were associated with heterozygous deletion of PARK2 at 6q25-q27 locus.[39] Parkin deficiency further diminished disease-free survival in infrared-irradiated mice without increasing tumour incidence rate, suggesting that parkin deficiencies increase susceptibility to tumour-promoting events, rather than initiating tumour formation.[10] Similarly, chromosomal breaks in PARK2 suppressed expression of afadin scaffold protein in breast cancer, thereby comprising epithelial integrity, enhancing metastatic potential and worsening overall prognosis.[40] Haploinsufficient PARK2 expression, either due to reduced copy number or DNA hypermethylation, was further detected in spontaneous colorectal cancer where it accelerated all stages of intestinal adenoma development in mouse models.[41] Parkin is therefore a potent modulator of tumour progression, without directly instigating tumourigenesis.
Interactions
Parkin (ligase) has been shown to interact with:
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000185345 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000023826 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (April 1998). "Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism". Nature. 392 (6676): 605–8. Bibcode:1998Natur.392..605K. doi:10.1038/33416. PMID 9560156. S2CID 4432261.
- ↑ Matsumine H, Yamamura Y, Hattori N, Kobayashi T, Kitada T, Yoritaka A, Mizuno Y (April 1998). "A microdeletion of D6S305 in a family of autosomal recessive juvenile parkinsonism (PARK2)". Genomics. 49 (1): 143–6. doi:10.1006/geno.1997.5196. PMID 9570960.
- ↑ Pickart CM, Eddins MJ (November 2004). "Ubiquitin: structures, functions, mechanisms". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1695 (1–3): 55–72. doi:10.1016/j.bbamcr.2004.09.019. PMID 15571809.
- 1 2 3 4 5 6 Seirafi M, Kozlov G, Gehring K (June 2015). "Parkin structure and function". The FEBS Journal. 282 (11): 2076–88. doi:10.1111/febs.13249. PMC 4672691. PMID 25712550.
- 1 2 3 4 5 Dawson TM, Dawson VL (2014). "The role of parkin in familial and sporadic Parkinson's disease". Movement Disorders. 25 (Suppl 1): S32-9. doi:10.1002/mds.22798. PMC 4115293. PMID 20187240.
- 1 2 3 4 Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z (2011). "Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect". Proceedings of the National Academy of Sciences of the United States of America. 108 (39): 16259–64. Bibcode:2011PNAS..10816259Z. doi:10.1073/pnas.1113884108. PMC 3182683. PMID 21930938.
- ↑ Trempe JF, Sauvé V, Grenier K, Seirafi M, Tang MY, Ménade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K (June 2013). "Structure of parkin reveals mechanisms for ubiquitin ligase activation". Science. 340 (6139): 1451–5. Bibcode:2013Sci...340.1451T. doi:10.1126/science.1237908. PMID 23661642. S2CID 206548928.
- 1 2 3 Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K, Diep L, Zhang Z, Chiou S, Bova M, Artis DR, Yao N, Baker J, Yednock T, Johnston JA (2013). "Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases". Nature Communications. 4: 1982. Bibcode:2013NatCo...4.1982R. doi:10.1038/ncomms2982. PMC 3709503. PMID 23770887.
- 1 2 3 Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (June 2014). "Ubiquitin is phosphorylated by PINK1 to activate parkin". Nature. 510 (7503): 162–6. Bibcode:2014Natur.510..162K. doi:10.1038/nature13392. PMID 24784582. S2CID 4390259.
- 1 2 Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N (July 2013). "Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation". The Journal of Biological Chemistry. 288 (30): 22019–32. doi:10.1074/jbc.M113.467530. PMC 3724655. PMID 23754282.
- ↑ Wauer T, Komander D (July 2013). "Structure of the human Parkin ligase domain in an autoinhibited state". The EMBO Journal. 32 (15): 2099–112. doi:10.1038/emboj.2013.125. PMC 3730226. PMID 23727886.
- 1 2 Olszewska, Diana Angelika; Lynch, Tim (2015). "Will Crystal Parkin Help in Understanding the Future of Parkinson’s Disease?". Frontiers in Neurology. 6: 35. doi:10.3389/fneur.2015.00035. PMC 4338761. PMID 25759682.
- 1 2 3 Durcan TM, Fon EA (May 2015). "The three 'P's of mitophagy: PARKIN, PINK1, and post-translational modifications". Genes & Development. 29 (10): 989–99. doi:10.1101/gad.262758.115. PMC 4441056. PMID 25995186.
- ↑ Youle RJ, van der Bliek AM (August 2012). "Mitochondrial fission, fusion, and stress". Science. 337 (6098): 1062–5. Bibcode:2012Sci...337.1062Y. doi:10.1126/science.1219855. PMC 4762028. PMID 22936770.
- ↑ Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (January 2008). "Fission and selective fusion govern mitochondrial segregation and elimination by autophagy". The EMBO Journal. 27 (2): 433–46. doi:10.1038/sj.emboj.7601963. PMC 2234339. PMID 18200046.
- 1 2 Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (April 2013). "Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization". Nature. 496 (7445): 372–6. Bibcode:2013Natur.496..372S. doi:10.1038/nature12043. PMC 3641819. PMID 23503661.
- 1 2 Narendra D, Walker JE, Youle R (November 2012). "Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism". Cold Spring Harbor Perspectives in Biology. 4 (11): a011338. doi:10.1101/cshperspect.a011338. PMC 3536340. PMID 23125018.
- ↑ Shlevkov E, Kramer T, Schapansky J, LaVoie MJ, Schwarz TL (October 2016). "Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility". Proceedings of the National Academy of Sciences of the United States of America. 113 (41): E6097–E6106. doi:10.1073/pnas.1612283113. PMC 5068282. PMID 27679849.
- ↑ Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (February 2010). "PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1". Nature Cell Biology. 12 (2): 119–31. doi:10.1038/ncb2012. PMID 20098416. S2CID 26096413.
- ↑ Aleksaniants GD (2013). "[Use of balneo-, peloid- and centimeter-wave therapy in the complex treatment of patients with circumscribed scleroderma]". Vestnik Dermatologii I Venerologii. 32 (6): 58–60. doi:10.1038/emboj.2013.70. PMC 3630365. PMID 23531882.
- ↑ Müller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, Lichtenthaler SF, Motori E, Hrelia S, Wurst W, Trümbach D, Langer T, Krappmann D, Dittmar G, Tatzelt J, Winklhofer KF (March 2013). "The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO". Molecular Cell. 49 (5): 908–21. doi:10.1016/j.molcel.2013.01.036. PMID 23453807.
- 1 2 3 da Costa CA, Sunyach C, Giaime E, West A, Corti O, Brice A, Safe S, Abou-Sleiman PM, Wood NW, Takahashi H, Goldberg MS, Shen J, Checler F (November 2009). "Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson's disease". Nature Cell Biology. 11 (11): 1370–5. doi:10.1038/ncb1981. PMC 2952934. PMID 19801972.
- ↑ Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM (June 2006). "p53 regulates mitochondrial respiration". Science. 312 (5780): 1650–3. Bibcode:2006Sci...312.1650M. doi:10.1126/science.1126863. PMID 16728594. S2CID 36668814.
- ↑ Poorkaj P, Nutt JG, James D, Gancher S, Bird TD, Steinbart E, Schellenberg GD, Payami H (August 2004). "parkin mutation analysis in clinic patients with early-onset Parkinson [corrected] disease". American Journal of Medical Genetics. Part A. 129A (1): 44–50. doi:10.1002/ajmg.a.30157. PMID 15266615. S2CID 85058092.
- ↑ Lohmann E, Periquet M, Bonifati V, Wood NW, De Michele G, Bonnet AM, Fraix V, Broussolle E, Horstink MW, Vidailhet M, Verpillat P, Gasser T, Nicholl D, Teive H, Raskin S, Rascol O, Destée A, Ruberg M, Gasparini F, Meco G, Agid Y, Durr A, Brice A (August 2003). "How much phenotypic variation can be attributed to parkin genotype?". Annals of Neurology. 54 (2): 176–85. doi:10.1002/ana.10613. PMID 12891670. S2CID 6411438.
- ↑ Ishikawa A, Takahashi H (November 1998). "Clinical and neuropathological aspects of autosomal recessive juvenile parkinsonism". Journal of Neurology. 245 (11 Suppl 3): P4-9. doi:10.1007/pl00007745. PMID 9808334. S2CID 28670790.
- ↑ Keeney PM, Xie J, Capaldi RA, Bennett JP (May 2006). "Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled". The Journal of Neuroscience. 26 (19): 5256–64. doi:10.1523/JNEUROSCI.0984-06.2006. PMC 6674236. PMID 16687518.
- ↑ Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (May 2006). "High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease". Nature Genetics. 38 (5): 515–7. doi:10.1038/ng1769. PMID 16604074. S2CID 13956928.
- ↑ Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T (July 2000). "Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase". Nature Genetics. 25 (3): 302–5. doi:10.1038/77060. PMID 10888878. S2CID 8135537.
- 1 2 Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM (October 2001). "Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease". Nature Medicine. 7 (10): 1144–50. doi:10.1038/nm1001-1144. PMID 11590439. S2CID 12487644.
- ↑ Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL, Dawson TM, Lim KL (February 2008). "Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases". Human Molecular Genetics. 17 (3): 431–9. doi:10.1093/hmg/ddm320. PMID 17981811.
- ↑ Corti O, Hampe C, Koutnikova H, Darios F, Jacquier S, Prigent A, Robinson JC, Pradier L, Ruberg M, Mirande M, Hirsch E, Rooney T, Fournier A, Brice A (June 2003). "The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: linking protein biosynthesis and neurodegeneration". Human Molecular Genetics. 12 (12): 1427–37. doi:10.1093/hmg/ddg159. PMID 12783850.
- ↑ Ko HS, Kim SW, Sriram SR, Dawson VL, Dawson TM (June 2006). "Identification of far upstream element-binding protein-1 as an authentic Parkin substrate". The Journal of Biological Chemistry. 281 (24): 16193–6. doi:10.1074/jbc.C600041200. PMID 16672220.
- ↑ Hattori N, Matsumine H, Asakawa S, Kitada T, Yoshino H, Elibol B, Brookes AJ, Yamamura Y, Kobayashi T, Wang M, Yoritaka A, Minoshima S, Shimizu N, Mizuno Y (August 1998). "Point mutations (Thr240Arg and Gln311Stop) [correction of Thr240Arg and Ala311Stop] in the Parkin gene". Biochemical and Biophysical Research Communications. 249 (3): 754–8. doi:10.1006/bbrc.1998.9134. PMID 9731209.
- ↑ Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA (January 2010). "Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies". Nature Genetics. 42 (1): 77–82. doi:10.1038/ng.491. PMC 4002225. PMID 19946270.
- ↑ Letessier A, Garrido-Urbani S, Ginestier C, Fournier G, Esterni B, Monville F, Adélaïde J, Geneix J, Xerri L, Dubreuil P, Viens P, Charafe-Jauffret E, Jacquemier J, Birnbaum D, Lopez M, Chaffanet M (January 2007). "Correlated break at PARK2/FRA6E and loss of AF-6/Afadin protein expression are associated with poor outcome in breast cancer". Oncogene. 26 (2): 298–307. doi:10.1038/sj.onc.1209772. PMID 16819513.
- ↑ Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, Arends MJ (August 2010). "PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice". Proceedings of the National Academy of Sciences of the United States of America. 107 (34): 15145–50. Bibcode:2010PNAS..10715145P. doi:10.1073/pnas.1009941107. PMC 2930574. PMID 20696900.
- ↑ Choi P, Golts N, Snyder H, Chong M, Petrucelli L, Hardy J, Sparkman D, Cochran E, Lee JM, Wolozin B (September 2001). "Co-association of parkin and alpha-synuclein". NeuroReport. 12 (13): 2839–43. doi:10.1097/00001756-200109170-00017. PMID 11588587. S2CID 83941655.
- 1 2 Kawahara K, Hashimoto M, Bar-On P, Ho GJ, Crews L, Mizuno H, Rockenstein E, Imam SZ, Masliah E (March 2008). "alpha-Synuclein aggregates interfere with Parkin solubility and distribution: role in the pathogenesis of Parkinson disease". The Journal of Biological Chemistry. 283 (11): 6979–87. doi:10.1074/jbc.M710418200. PMID 18195004.
- ↑ Fallon L, Moreau F, Croft BG, Labib N, Gu WJ, Fon EA (January 2002). "Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain". The Journal of Biological Chemistry. 277 (1): 486–91. doi:10.1074/jbc.M109806200. PMID 11679592.
- 1 2 Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A (March 2003). "Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity". Neuron. 37 (5): 735–49. doi:10.1016/s0896-6273(03)00084-9. PMID 12628165. S2CID 17024826.
- 1 2 3 4 Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R (July 2002). "CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity". Molecular Cell. 10 (1): 55–67. doi:10.1016/s1097-2765(02)00583-x. PMID 12150907.
- ↑ Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R (June 2001). "An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin". Cell. 105 (7): 891–902. doi:10.1016/s0092-8674(01)00407-x. PMID 11439185. S2CID 721363.
- ↑ Corti O, Hampe C, Koutnikova H, Darios F, Jacquier S, Prigent A, Robinson JC, Pradier L, Ruberg M, Mirande M, Hirsch E, Rooney T, Fournier A, Brice A (June 2003). "The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: linking protein biosynthesis and neurodegeneration". Human Molecular Genetics. 12 (12): 1427–37. doi:10.1093/hmg/ddg159. PMID 12783850.
- ↑ Fukae J, Sato S, Shiba K, Sato K, Mori H, Sharp PA, Mizuno Y, Hattori N (February 2009). "Programmed cell death-2 isoform1 is ubiquitinated by parkin and increased in the substantia nigra of patients with autosomal recessive Parkinson's disease". FEBS Letters. 583 (3): 521–5. doi:10.1016/j.febslet.2008.12.055. hdl:1721.1/96274. PMID 19146857. S2CID 7121769.
- ↑ Choi P, Snyder H, Petrucelli L, Theisler C, Chong M, Zhang Y, Lim K, Chung KK, Kehoe K, D'Adamio L, Lee JM, Cochran E, Bowser R, Dawson TM, Wolozin B (October 2003). "SEPT5_v2 is a parkin-binding protein". Brain Research. Molecular Brain Research. 117 (2): 179–89. doi:10.1016/s0169-328x(03)00318-8. PMID 14559152.
- ↑ Liu M, Aneja R, Sun X, Xie S, Wang H, Wu X, Dong JT, Li M, Joshi HC, Zhou J (December 2008). "Parkin regulates Eg5 expression by Hsp70 ubiquitination-dependent inactivation of c-Jun NH2-terminal kinase". The Journal of Biological Chemistry. 283 (51): 35783–8. doi:10.1074/jbc.M806860200. PMID 18845538.
- ↑ Huynh DP, Scoles DR, Nguyen D, Pulst SM (October 2003). "The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI". Human Molecular Genetics. 12 (20): 2587–97. doi:10.1093/hmg/ddg269. PMID 12925569.
- ↑ Yu F, Zhou J (July 2008). "Parkin is ubiquitinated by Nrdp1 and abrogates Nrdp1-induced oxidative stress". Neuroscience Letters. 440 (1): 4–8. doi:10.1016/j.neulet.2008.05.052. PMID 18541373. S2CID 2169911.
Further reading
- Saito M, Matsumine H, Tanaka H, Ishikawa A, Matsubayashi S, Hattori Y, Mizuno Y, Tsuji S (January 1997). "[Clinical characteristics and linkage analysis of autosomal recessive form of juvenile parkinsonism(AR-JP)]". Nihon Rinsho. Japanese Journal of Clinical Medicine. 55 (1): 83–8. PMID 9014427.
- Fishman PS, Oyler GA (July 2002). "Significance of the parkin gene and protein in understanding Parkinson's disease". Current Neurology and Neuroscience Reports. 2 (4): 296–302. doi:10.1007/s11910-002-0004-7. PMID 12044248. S2CID 21068281.
- Takahashi R (June 2002). "[Function of Parkin]". Seikagaku. The Journal of Japanese Biochemical Society. 74 (6): 471–6. PMID 12138708.
- West AB, Maidment NT (March 2004). "Genetics of parkin-linked disease". Human Genetics. 114 (4): 327–36. doi:10.1007/s00439-003-1074-6. PMID 14727181. S2CID 23264061.
- Mata IF, Lockhart PJ, Farrer MJ (April 2004). "Parkin genetics: one model for Parkinson's disease". Human Molecular Genetics. 13 Spec No 1 (90001): R127-33. doi:10.1093/hmg/ddh089. PMID 14976155.
- Baptista MJ, Cookson MR, Miller DW (February 2004). "Parkin and alpha-synuclein: opponent actions in the pathogenesis of Parkinson's disease". The Neuroscientist. 10 (1): 63–72. doi:10.1177/1073858403260392. PMID 14987449. S2CID 84671340.
- Kahle PJ, Haass C (July 2004). "How does parkin ligate ubiquitin to Parkinson's disease?". EMBO Reports. 5 (7): 681–5. doi:10.1038/sj.embor.7400188. PMC 1299099. PMID 15229644.
- Pankratz N, Foroud T (April 2004). "Genetics of Parkinson disease". NeuroRx. 1 (2): 235–42. doi:10.1602/neurorx.1.2.235. PMC 534935. PMID 15717024.
- Suzuki H (September 2006). "Protein-protein interactions in the mammalian brain". The Journal of Physiology. 575 (Pt 2): 373–7. doi:10.1113/jphysiol.2006.115717. PMC 1819454. PMID 16840513.
- Hattori N, Machida Y, Sato S, Noda K, Iijima-Kitami M, Kubo S, Mizuno Y (2006). "Molecular mechanisms of nigral neurodegeneration in Park2 and regulation of parkin protein by other proteins". Journal of Neural Transmission. Supplementum. Journal of Neural Transmission. Supplementa. 70 (70): 205–8. doi:10.1007/978-3-211-45295-0_31. ISBN 978-3-211-28927-3. PMID 17017530.
- Matsumine H, Saito M, Shimoda-Matsubayashi S, Tanaka H, Ishikawa A, Nakagawa-Hattori Y, Yokochi M, Kobayashi T, Igarashi S, Takano H, Sanpei K, Koike R, Mori H, Kondo T, Mizutani Y, Schäffer AA, Yamamura Y, Nakamura S, Kuzuhara S, Tsuji S, Mizuno Y (March 1997). "Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27". American Journal of Human Genetics. 60 (3): 588–96. PMC 1712507. PMID 9042918.
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (April 1998). "Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism". Nature. 392 (6676): 605–8. Bibcode:1998Natur.392..605K. doi:10.1038/33416. PMID 9560156. S2CID 4432261.
- Matsumine H, Yamamura Y, Hattori N, Kobayashi T, Kitada T, Yoritaka A, Mizuno Y (April 1998). "A microdeletion of D6S305 in a family of autosomal recessive juvenile parkinsonism (PARK2)". Genomics. 49 (1): 143–6. doi:10.1006/geno.1997.5196. PMID 9570960.
- Tassin J, Dürr A, de Broucker T, Abbas N, Bonifati V, De Michele G, Bonnet AM, Broussolle E, Pollak P, Vidailhet M, De Mari M, Marconi R, Medjbeur S, Filla A, Meco G, Agid Y, Brice A (July 1998). "Chromosome 6-linked autosomal recessive early-onset Parkinsonism: linkage in European and Algerian families, extension of the clinical spectrum, and evidence of a small homozygous deletion in one family. The French Parkinson's Disease Genetics Study Group, and the European Consortium on Genetic Susceptibility in Parkinson's Disease". American Journal of Human Genetics. 63 (1): 88–94. doi:10.1086/301934. PMC 1377254. PMID 9634531.
- Hattori N, Matsumine H, Asakawa S, Kitada T, Yoshino H, Elibol B, Brookes AJ, Yamamura Y, Kobayashi T, Wang M, Yoritaka A, Minoshima S, Shimizu N, Mizuno Y (August 1998). "Point mutations (Thr240Arg and Gln311Stop) [correction of Thr240Arg and Ala311Stop] in the Parkin gene". Biochemical and Biophysical Research Communications. 249 (3): 754–8. doi:10.1006/bbrc.1998.9134. PMID 9731209.
- Lücking CB, Abbas N, Dürr A, Bonifati V, Bonnet AM, de Broucker T, De Michele G, Wood NW, Agid Y, Brice A (October 1998). "Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European Consortium on Genetic Susceptibility in Parkinson's Disease and the French Parkinson's Disease Genetics Study Group". Lancet. 352 (9137): 1355–6. doi:10.1016/S0140-6736(05)60746-5. PMID 9802278. S2CID 44409075.
- Abbas N, Lücking CB, Ricard S, Dürr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Böhme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A (April 1999). "A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease". Human Molecular Genetics. 8 (4): 567–74. doi:10.1093/hmg/8.4.567. PMID 10072423.
- Sunada Y, Saito F, Matsumura K, Shimizu T (October 1998). "Differential expression of the parkin gene in the human brain and peripheral leukocytes". Neuroscience Letters. 254 (3): 180–2. doi:10.1016/S0304-3940(98)00697-1. PMID 10214987. S2CID 32794960.
- Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, Mizuno Y (May 1999). "Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients". Annals of Neurology. 45 (5): 668–72. doi:10.1002/1531-8249(199905)45:5<668::AID-ANA19>3.0.CO;2-Z. PMID 10319893.
Vanjski linkovi
- GeneReviews/NCBI/NIH/UW entry on Parkin Type of Juvenile Parkinson Disease
- parkin+protein at the US National Library of Medicine Medical Subject Headings (MeSH)
PDB gallery | |
|---|---|
|