Paroxysmal kinesigenic choreoathetosis
| Paroxysmal kinesigenic choreoathetosis | |
|---|---|
| Other names | Familial PKD |
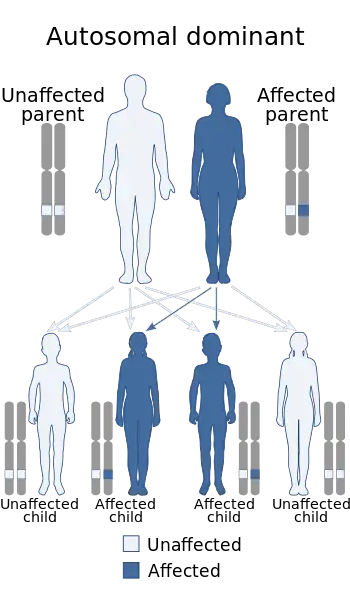 | |
| Paroxysmal kinesigenic choreoathetosis is inherited in an autosomal dominant manner | |
| Specialty | Neurology |
Paroxysmal kinesigenic choreoathetosis (PKC) also called paroxysmal kinesigenic dyskinesia (PKD) is a hyperkinetic movement disorder characterized by attacks of involuntary movements, which are triggered by sudden voluntary movements. The number of attacks can increase during puberty and decrease in a person's 20s to 30s. Involuntary movements can take many forms such as ballism, chorea or dystonia and usually only affect one side of the body or one limb in particular. This rare disorder only affects about 1 in 150,000 people,[1] with PKD accounting for 86.8% of all the types of paroxysmal dyskinesias,[2] and occurs more often in males than females. There are two types of PKD, primary and secondary. Primary PKD can be further broken down into familial and sporadic. Familial PKD, which means the individual has a family history of the disorder, is more common, but sporadic cases are also seen.[3] Secondary PKD can be caused by many other medical conditions such as multiple sclerosis (MS), stroke, pseudohypoparathyroidism,[4] hypocalcemia, hypoglycemia, hyperglycemia,[3] central nervous system trauma, or peripheral nervous system trauma.[5] PKD has also been linked with infantile convulsions and choreoathetosis (ICCA) syndrome, in which patients have afebrile seizures during infancy (benign familial infantile epilepsy) and then develop paroxysmal choreoathetosis later in life.[6] This phenomenon is actually quite common, with about 42% of individuals with PKD reporting a history of afebrile seizures as a child.[6]
Genetics
Paroxysmal kinesigenic dyskinesias are often inherited in an autosomal dominant fashion and several genes have now been identified where mutations can cause this disease. The genes typically code for proteins known to be involved in synaptic transmission, ion channels or ion transporters.[7] The first gene to be identified was the PRRT2 gene on chromosome 16, found in 2011 to be the cause of the disease in some patients.[8] The mutations in this gene included a nonsense mutation identified in the genome of one family and an insertion mutation identified in the genome of another family.[9] Researchers found PRRT2 mutations in 10 of 29 sporadic cases affected with PKD, thus suggests PRRT2 is the gene mutated in a subset of PKD and PKD is genetically heterogeneous.[10] Later reports have identified the genes SCN8A, CHRNA4, and SLC16A2 as further causes of PKD.[7]
Pathophysiology
The pathophysiology of PKD is not fully explained. A few mechanisms have been suggested thus far:
- GABA dysregulation
- Abnormal breakdown of dopamine in the basal ganglia
- Dysfunction of the substantia nigra[11]
- A form of epilepsy[2]
Multiple methods are being used to study the potential brain abnormalities of individuals with PKD compared with “normal” individuals. These methods include SPECT studies, fMRI studies, and diffusion tensor imaging. The main problem with many of the studies concerned with the pathophysiology of the disorder is the small sample size. Because the studies normally only include about 7-10 patients with PKD, the results cannot be generalized to the entire population of patients. However, the studies do bring up possibilities for further study.
SPECT studies
In a study by Joo et al., the researchers performed interictal studies, meaning they scanned the patient's brain between attacks to find an underlying abnormality, rather than ictal scans, which look at the abnormalities that present themselves during an attack.[11] The researchers found interictally decreased cerebral blood flow in the posterior parts of the bilateral caudate nucleus.[11] However, the literature does state that although this could be a cause of PKD, it could also be a result of PKD.[11] Another SPECT study showed an increase in the cerebral blood flow in the left posterior thalamus in a PKD patient during an attack.[12] The researchers also subtracted the ictal from the postictal scans, and saw increased blood flow in the thalamus. They ultimately suggested that hyperactive blood flow in this area could be causing the pathophysiology of PKD. This study, however, was only performed on one patient, and would need to be replicated many more times in order to be generalized to the population of PKD patients. Other SPECT studies have been cited showing hyperactivity in the basal ganglia.[13]
fMRI studies
In a study by Zhou et al.,[14] the researchers performed fMRI studies on PKD patients, and analyzed the differences between the amplitude low frequency fluctuations (ALFF) of the patients. They found that the left postcentral gyrus and the bilateral putamen had increased ALFF in PKD patients.[14] The researchers concluded that the hyperactivity in these regions suggested that there is a dysfunction in the basal ganglia-thalamo-cortical circuit in PKD. This circuit is part of the motor control circuit in the brain, making it a reasonable place for abnormality in a movement disorder, but again, researchers are still unsure of the role these differences they found play in the disease pathology.
Diffusion tensor imaging
Diffusion tensor imaging (DTI) displays physical alterations in the brain that may not be seen on regular MRI.[13] In one study researchers found that some of the patients had abnormalities in their thalamus. However, this does not prove that all patients have abnormalities in their thalamus. Other cases are cited, including a patient who developed a similar paroxysmal dyskinesia after a thalamic infarction,[13] implicating that an abnormality in the thalamus of individuals could contribute to PKD. It is not fully known, however, what role a thalamic abnormality plays in the disease pathophysiology.
Diagnosis
Paroxysmal kinesigenic dyskinesia is diagnosed using a strict set of guidelines. These criteria were studied and confirmed by Bruno et al. in a study of 121 individuals with PKD.[3] The age at onset is between 1 and 20 years old. The attacks of involuntary movements last less than one minute and have a known trigger, usually a sudden voluntary movement. For example, if a PKD patient stands up or begins walking after being sedentary for a period of time, or a person goes from a walk to a run, it can trigger an attack. Persons with PKD do not lose consciousness during attacks and have a full memory of the entire attack. Lastly, people with the disorder have a good response to medication and are usually prescribed anticonvulsants. The study also found that patients with familial PKD exhibit symptoms that follow the diagnostic criteria closely, while sporadic PKD individuals may deviate slightly.[5] Prior to criteria for diagnosis being set out, many patients with PKD were often diagnosed with some form of epilepsy. Many patients also experience an aura, similar to those experienced with epilepsy, preceding their attacks. Some patients describe it as a tingling sensation in the affected limb or “butterflies in their stomach.” Some individuals also have precipitants, such as stress and anxiety, that make it more likely for attacks to occur.
The above diagnostic criteria also set PKD apart from the other paroxysmal dyskinesias, which include paroxysmal nonkinesigenic dyskinesia (PNKD) and paroxysmal exercise-induced dyskinesia (PED). While PKD attacks last less than one minute, PNKD attacks last a few minutes to a few hours, and as the name suggests, the attacks do not occur because of a sudden voluntary movement like PKD.[5] Additionally, PKD can almost always be managed with drug therapy, while PNKD is not as responsive to anticonvulsants. PED, on the other hand, separates itself from PKD in that it is caused by prolonged exercise. Attacks from PED will cease soon after exercise is stopped.[5]
Treatment
Almost all patients respond positively to antiepileptic (anticonvulsant) drugs. One of the drugs most often mentioned in the literature is carbamazepine, and is the most widely used drug for treating PKD. Other anticonvulsants like valproic acid, phenytoin and clonazepam are common alternatives. Other categories of drugs have also been used, such as dopamine affecting drugs like Levodopa or Tetrabenazine.[5] Individuals with the disorder can also modify their behavior to lessen their attacks without the influence of drug therapy. For example, decreasing stress to avoid precipitants can help patients decrease the number of attacks. In addition, avoiding any sudden movements can also prevent an attack. In order to prevent an attack, some individuals use their auras as a warning, while others purposefully perform slow gestures or movements prior to a triggering movement.[2] Many, if not most, individuals end up growing out of the attacks with age, even without medicinal therapy, but some patients will go back to having attacks after a period of remission.[3] In regards to secondary PKD, treatment of the primary condition can lessen the PKD attacks in those individuals.[5]
History
A movement order similar to PKD first mentioned in research literature in 1940 by Mount and Reback. They described a disorder consisting of attacks of involuntary movements but unlike PKD, the attacks lasted minutes to hours and were found to be caused by alcohol or caffeine intake.[15] They named it paroxysmal dystonic choreoathetosis. Kertesz later described another new movement disorder in 1967. He described a disorder that was induced by sudden movements, and responded to anticonvulsants, naming it paroxysmal kinesigenic choreoathetosis. Finally in a review in 1995 Demirkiran and Jankovic stated the disease should be called paroxysmal kinesigenic dyskinesia instead, pointing out that the attacks could manifest as any form of dyskinesia, not just choreoathetosis.[15]
See also
References
- ↑ Khan, W. U.; Staios, G.; Rana, A. Q. (2010). "Paroxysmal kinesigenic dyskinesia in a mother and daughter". Acta Neurologica Belgica. 110 (2): 201–202. PMID 20873453.
- 1 2 3 Zhou, J. Q.; Zhou, L. M.; Fang, Z. Y.; Wang, Q.; Chen, Z. Y.; Yang, L. B.; Chen, S. D.; Cai, X. D. (2011). "Analyzing clinical and electrophysiological characteristics of Paroxysmal Dyskinesia". Journal of Research in Medical Sciences. 16 (1): 110–114. PMC 3063430. PMID 21448393.
- 1 2 3 4 Bruno, M. K.; Hallett, M.; Gwinn-Hardy, K.; Sorensen, B.; Considine, E.; Tucker, S.; Lynch, D. R.; Mathews, K. D.; Swoboda, K. J.; Harris, J.; Soong, B. W.; Ashizawa, T.; Jankovic, J.; Renner, D.; Fu, Y. H.; Ptacek, L. J. (2004). "Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia: New diagnostic criteria". Neurology. 63 (12): 2280–2287. doi:10.1212/01.WNL.0000147298.05983.50. PMID 15623687.
- ↑ Thomas, K. P.; Muthugovindan, D.; Singer, H. S. (2010). "Paroxysmal Kinesigenic Dyskinesias and Pseudohypo-parathyroidism Type Ib". Pediatric Neurology. 43 (1): 61–64. doi:10.1016/j.pediatrneurol.2010.03.012. PMID 20682207.
- 1 2 3 4 5 6 Mehta, S. H.; Morgan, J. C.; Sethi, K. D. (2009). "Paroxysmal dyskinesias". Current Treatment Options in Neurology. 11 (3): 170–178. doi:10.1007/s11940-009-0020-x. PMID 19364451.
- 1 2 Weber, Y. G.; Lerche, H. (2009). "Genetics of paroxysmal dyskinesias". Current Neurology and Neuroscience Reports. 9 (3): 206–211. doi:10.1007/s11910-009-0031-8. PMID 19348709.
- 1 2 Papandreou A, Danti FR, Spaull R, Leuzzi V, Mctague A, Kurian MA (February 2020). "The expanding spectrum of movement disorders in genetic epilepsies". Developmental Medicine and Child Neurology. 62 (2): 178–191. doi:10.1111/dmcn.14407. PMID 31784983.
- ↑ Chen, W. J.; Lin, Y.; Xiong, Z. Q.; Wei, W.; Ni, W.; Tan, G. H.; Guo, S. L.; He, J.; Chen, Y. F.; Zhang, Q. J.; Li, H. F.; Lin, Y.; Murong, S. X.; Xu, J.; Wang, N.; Wu, Z. Y. (2011). "Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia". Nature Genetics. 43 (12): 1252–1255. doi:10.1038/ng.1008. PMID 22101681.
- ↑ Wang, J. -L.; Cao, L.; Li, X. -H.; Hu, Z. -M.; Li, J. -D.; Zhang, J. -G.; Liang, Y.; San-a; Li, N.; Chen, S. -Q.; Guo, J. -F.; Jiang, H.; Shen, L.; Zheng, L.; Mao, X.; Yan, W. -Q.; Zhou, Y.; Shi, Y. -T.; Ai, S. -X.; Dai, M. -Z.; Zhang, P.; Xia, K.; Chen, S. -D.; Tang, B. -S. (2011). "Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias". Brain. 134 (12): 3493–3501. doi:10.1093/brain/awr289. PMC 3235563. PMID 22120146.
- ↑ Li J, Zhu X, Wang X et al. J Med Genet. 2012 Feb;49(2):76-8. Epub 2011 Nov 30. Targeted genomic sequencing identifies PRRT2 mutations as a cause of paroxysmal kinesigenic choreoathetosis.
- 1 2 3 4 Joo, E. Y.; Hong, S. B.; Tae, W. S.; Kim, J. H.; Han, S. J.; Seo, D. W.; Lee, K. H.; Kim, M. H.; Kim, S.; Lee, M. H.; Kim, B. T. (2005). "Perfusion abnormality of the caudate nucleus in patients with paroxysmal kinesigenic choreoathetosis". European Journal of Nuclear Medicine and Molecular Imaging. 32 (10): 1205–1209. doi:10.1007/s00259-005-1814-z. PMID 15948007.
- ↑ Shirane, S.; Sasaki, M; Kogure, D; Matsuda, H; Hashimoto, T (2001). "Increased ictal perfusion of the thalamus in paroxysmal kinesigenic dyskinesia". Journal of Neurology, Neurosurgery & Psychiatry. 71 (3): 408–410. doi:10.1136/jnnp.71.3.408. PMC 1737540. PMID 11511723.
- 1 2 3 Zhou, B.; Chen, Q.; Gong, Q.; Tang, H.; Zhou, D. (2009). "The thalamic ultrastructural abnormalities in paroxysmal kinesigenic choreoathetosis: A diffusion tensor imaging study". Journal of Neurology. 257 (3): 405–409. doi:10.1007/s00415-009-5334-9. PMID 20012544.
- 1 2 Zhou, B.; Chen, Q.; Zhang, Q.; Chen, L.; Gong, Q.; Shang, H.; Tang, H.; Zhou, D. (2010). "Hyperactive putamen in patients with paroxysmal kinesigenic choreoathetosis: A resting-state functional magnetic resonance imaging study". Movement Disorders. 25 (9): 1226–1231. doi:10.1002/mds.22967. PMID 20629125.
- 1 2 Bhatia, K. P. (2011). "Paroxysmal dyskinesias". Movement Disorders. 26 (6): 1157–1165. doi:10.1002/mds.23765. PMID 21626559.