Passive antibody therapy
Passive antibody therapy, also called serum therapy, is a subtype of passive immunotherapy that administers antibodies (same as immunoglobin) to target and kill pathogens or cancer cells.[1] It is designed to draw support from foreign antibodies that are donated from a person, extracted from animals, or made in the laboratory to elicit an immune response instead of relying on the innate immune system to fight disease. It has a long history from the 18th century for treating infectious diseases and is now a common cancer treatment. The mechanism of actions include: antagonistic and agonistic reaction, complement-dependent cytotoxicity (CDC), and antibody-dependent cellular cytotoxicity (ADCC).
History
.png.webp)
Passive antibody therapy was first propounded by Emil von Behring and Shibasaburo Kitasato in 1890 to treat diphtheria after the observation of immunization in rabbits after injecting serum from tetanus-immunized rabbits. Later in 1891, Paul Ehrlich joined Behring's and Kitasato's research to ameliorate immunizability from lethal toxins. This established the basis of antibody immunotherapy. With the ideology of using antibody serum to treat infectious diseases, the three scientists standardized serum production in dairy cows and merchandised serum vaccines for tetanus and diphtheria.
The prevalence of serum therapy surged in the early 19th century. When the H1N1 influenza pandemic (Spanish flu) struck the US and Europe, serum containing antibodies from recovered patients are prevalently injected into patients. With proven therapeutic effects, the applications expanded to other viral and bacterial infections, such as pneumococcus, meningococcus, and rabies, despite an unknown underlying mechanism. Yet, severe anaphylactic reactions and hypersensitivity were common, ergo, serum therapy was pulled out from the market in the 1940s. The resurrection of antibody immunotherapy contributed to Cesar Milstein and Georges J. F. Kohler, who manifested the mass production of pure monoclonal antibodies with limited adverse effects in 1975. Since then, passive antibody therapy has become prevailed as cancer therapeutics and viral treatments.
Classification of passive immunity therapy
| Monoclonal Antibodies (mAb) | Polyclonal Antibodies (pAb) | |
|---|---|---|
| Characteristics | A single type of antibodies that recognize one epitope of an antigen | Contain a mixture of antibodies that can recognize multiple epitopes on a single type of antigen |
| Manufacture process | Create hybridoma: isolate B lymphocytes from animal spleen and fuse with myeloma cell line | Directly collect serum from immunized animals and purify the solution to remove other serum proteins |
| Advantages | - high homogeneity (low batch-to-batch variability)
- high specificity and low cross-reactivity - high sensitivity |
- inexpensive to produce
- high overall affinity - bind to target antigen quicker |
| Disadvantages | - expensive to produce
- long production time - overall affinity is lower than polyclonal antibodies |
- high batch-to-batch variability
- low specificity and high cross-reactivity |
Monoclonal antibodies (mAb)
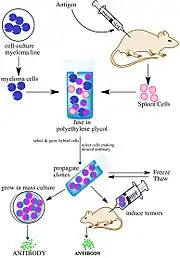
Monoclonal antibodies are manufactured ex vivo from a single B lymphocyte. Serum from immunized animals or humans is first extracted and purified to collect B lymphocytes from the spleen, which are then fused with plasma cell myeloma. After culturing the fused myeloma cell lines, the colonies are selected with the antigens: positive colonies with suitable antibodies can bind to the epitope of the antigen and kill pathogens, whereas colonies without targeted antibodies are eliminated.[2] Upon injection, these homogenous antibodies produced from a single B cell can target a specific epitope on the antigen.[3] The major advantage of using monoclonal antibodies is their specific action towards the target since it only contains one antibody binding site per se, it minimizes cross-reactivity (the activity that antibodies unintentionally bind to non-targeted antigens).[4] However, monoclonal antibodies also mean that overall affinity is lower owing to the limited ability to recognize different epitopes on the antigens, which may lead to incomplete elimination of pathogens and tumor cells. The production time and cost is high as well, limiting its generalizability and prevalence of usage.[5]
Polyclonal antibodies (pAb)
The process of manufacturing polyclonal antibodies is similar to that of monoclonal antibodies, which begins with inoculation of antigen conjugate into suitable animals, except multiple B lymphocytes are collected and cultured instead of a single B lymphocyte.[6] Production of polyclonal antibodies circumvents the procedure of ex vivo fabrication of hybridoma cell line and requires minimal purification. The manufacturing cost and time are wherefore reduced. Due to a heterogeneous origin, the antibodies express various subtypes of immunoglobulin against the antigen[3] which has an overall higher affinity and can better detect low-quantity antigens by targeting different epitopes on the antigen.[7] However, it also provokes an increased chance of non-specific reactivity because the antibodies might bind to non-diseases causing substances. In addition, as serum batch may contain various antibodies at different concentrations, it is laborious to corroborate the constituents of every batch.
Mechanism of Action
Since some patients fail to produce antibodies effectively and hence have poorer immune responses, passive antibody therapy can reinforce their immune system through the introduction of antibodies from donors. Antibodies are glycoproteins that are naturally produced by the immune system. Each antibody contains four polypeptides of Y shapes and has unique recognition sites of the targets, such as cell surface antigen, and transmembrane proteins on cancer cells and infectious organisms (viruses and bacteria). Upon binding to the antigen, antibodies trigger different cascades to neutralize toxins and kill the cells. There are three ways of action: antagonistic and agonistic reaction, complement-dependent cytotoxicity (CDC), and antibody-dependent cellular cytotoxicity (ADCC).
Antagonistic reaction (Neutralization) and Agonistic reaction
Antagonism by antibodies eliminates antigens by binding to the relevant Fc receptors or pathogens for disrupting the toxins from binding to the receptors. In cancers, tumor cells escape immune vigilance by binding to checkpoint proteins on immune cells for inhibiting immune signaling and downregulating the expression of major histocompatibility class I (MHC I).[8] Antagonistic antibodies, also called immune checkpoints inhibitors, obstruct the binding between cancer cells and immune checkpoints to antagonize cancer cells’ action and restore immune surveillance. Therefore, immune cells can recognize the surface antigens on the tumor cells to elicit immune responses. Examples of drugs that exploit such a mechanism include pembrolizumab and telimomab.
Apart from directing the inhibitory pathways, agonistic antibodies can target immunostimulatory receptors to elicit immune responses. Upon binding between cluster of differentiation proteins (CD proteins) and agonistic antibodies, antigen-presenting cells, such as dendritic cells, B lymphocytes and monocytes, are stimulated to secrete proinflammatory cytokines to remove pathogens and malignant cells. For example, the ligation of CD40 monoclonal antibodies and CD40 on tumor cells license antigen-presenting cells (predominantly dendritic cells) to increase the presentation of tumor-associated antigens (TAA) to local cytotoxic T lymphocytes to kill tumor cells.[8]
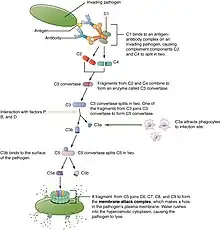
Complement-dependent Cytotoxicity (CDC) Pathway
Antibodies can also trigger the classical pathway – one of the three pathways of the complement cascade. Briefly, the C1 protein attaches to the pathogen surface and the antibody-antigen complex that culminates in the generation of C3 convertase, followed by the cleavage of C3 protein into C3a and C3b protein. C3a protein serves as an inflammation mediator to recruit phagocytes. On the other hand, C3 protein can opsonize pathogens and bind to C3 convertase to catalyze the formation of C5 convertase to produce C5a and C5b for terminal complement components assembly.[9] The formation of complement proteins (C3a, C3b, C5a, C5b, etc.) ultimately congregates into a membrane-attack complex to lyse the membrane of pathogens.[10]
In addition to the generation of complement proteins, C1 complex also induces the activation of B cells, monocytes, macrophages, and neutrophils to trigger immune responses, for instance, vasodilation and increased vascular permeability at the infection site.
Antibody-dependent Cellular Cytotoxicity (ADCC) Pathway
Several monoclonal antibody drugs such as Rituximab manipulate the ADCC pathway to eradicate cancer cells. Firstly, the Fab region of the antibodies (tip of the Y shaped-antibody) first binds to the epitope of the pathogens or the immunoreceptors of cancer cells; whereas the Fc region (stem of the antibody) binds to natural killer cells to phosphorylate the immunoreceptor. Several notable immunoreceptors for cancer treatment include epidermal growth factor receptor (EFGR), insulin-like growth factor receptor (IGFR), and cMET as they are commonly overexpressed in malignant cells. Phosphorylation of immunoreceptors triggers the release of cytotoxic granules from the natural killer cells which sequentially perturbs cell-cell signaling via the induction of the apoptotic cascade (Bax, Bad, etc.),[11] leading to cell death and enhancing the susceptibility of cancer cells to chemotherapy or radiotherapy.
Application on Cancer
.jpg.webp)
Passive antibody administration has become a widely approved cancer treatment following the development of monoclonal antibody (mAb). Since these antibodies originated from mice, they were wrought with problems of immunogenetics and poor abilities to induce an immune response in the human body, limiting their clinical applicability.[12] Later development of antibody engineering enabled the production of chimeric antibodies (antibodies with human Fc region and mouse Fab region), humanized antibodies (antibodies with replaced mouse complementarity-determining regions and human backbone), and full human antibodies (antibodies produced by transgenic mice), which satisfactorily addressed many of these problems and were suitable for the clinical cancer treatment.[13]
Cetuximab
Cetuximab (trade name: Erbitux ) is a recombinant chimeric monoclonal antibody designed to treat metastatic colorectal cancer and head and neck cancer.[14] In numerous cancers, the epidermal growth factor receptor (EGFR) is often inappropriately activated and overexpressed in cancer cells, leading to uncontrolled cell growth.[15] Cetuximab blocks ligand binding of EGFR and inhibits intracellular downstream events critical for tumor survival, thereby inducing tumor regression by reducing cell proliferation and increasing apoptosis.[15] It is administered by intravenous injection and is often prescribed in combination with radiotherapy or other chemotherapeutic regimens such as irinotecan. Since EGFR plays an essential role in maintaining skin integrity, one serious side effect of Cetuximab therapy is skin problems such as acne-like rash, skin drying and cracking due to the inhibition of EGFR.[14] Other serious side effects are severe allergic reactions and heart attacks.[16]
Rituximab
In 1997, the FDA approved rituximab (trade names: Rituxan, MabThera) as the first monoclonal antibody for clinical cancer treatment.[17] This drug directly targets CD20 that is found on the surface of both normal and malignant B cells and is indicated to treat blood cancers, such as non-Hodgkin's lymphoma (a type of B cell lymphoma) and chronic lymphocytic leukemia (B cell malignancy), and other conditions like rheumatoid arthritis and vasculitis.[18] The mechanism of action includes B-cell lysis by antibody-dependent cellular cytotoxicity (ADCC), and complement-dependent cytotoxicity (CDC).[19] Common side effects include itching, headache, nausea, diarrhea and low blood pressure.[20]
Brentuximab vedotin
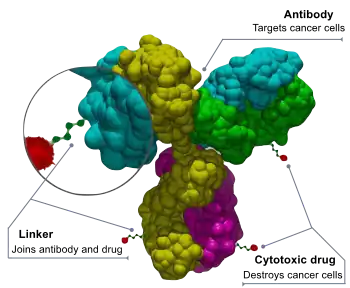
Brentuximab vedotin (trade name: Adcetris) is a CD30 targeted antibody-drug conjugate (ADC), i.e. a monoclonal antibody chemically linked to a drug, and is constructed by chimeric anit-CD30 IgG1 antibody (cAC10), monomethyl auristatin E (MMAE, a potent cytotoxic drug) and a linker that attaches MMAE covalently to cAC10.[21] The binding of brentuximab vedotin to tumor cells is followed by the release of cytotoxic drug MMAE, which destroys the microtubule network within the cell and induces cell apoptosis.[22] Brentuximab vedotin became the first antibody-drug conjugate approved by FDA to treat Hodgkin's lymphoma and anaplastic large-cell lymphoma in 2011.[23]
Application on infectious diseases
Palivizumab
In 1998, the FDA approved the medical use of palivizumab (trade name: Synagis), which is a humanized monoclonal antibody used as prophylaxis to prevent severe diseases caused by respiratory syncytial virus (RSV) - the leading cause of lower respiratory tract infections in infants.[24] The drug is recommended for infants with a high risk of RSV infection due to prematurity or diseases such as congenital heart disease (CHD) and bronchopulmonary dysplasia (BPD).[24] However, it will not treat children already infected with RSV. Injected intramuscularly, it binds to RSV fusion proteins to prevent RSV from binding to host cell receptors and uptake. Common side effects are diarrhea, vomiting, runny nose and other cold symptoms.[25]
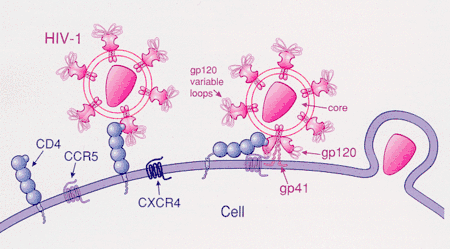
Ibalizumab
Ibalizumab (trade name: Trogarzo) is a new antiviral drug specifically for adults with HIV who have tried but developed resistant against other HIV therapies. The entry of HIV into human immune cells requires the binding of the GP120 (an envelope glycoprotein on the surface of HIV) to CD4 (a receptor on the immune cell surface) to induce the structural shift in the GP120.[26] Ibalizumab binds to the CD4 receptor to prevent the post-attachment conformational changes in CD4-HIV envelope GP120 complex and therefore hinders the viral entry into host cells and prevents the HIV-infected cells from infecting other normal cells.[27] The advantages of using Ibalizumab as HIV therapy are low toxicity, good drug resistance, high antiviral effect and synergy with other antiviral drugs.[27]
Perspective and future opportunities
Although passive administration of antibodies was used to treat infectious diseases 100 years ago, there are only a few therapeutic antibodies approved for the clinical treatments of infectious diseases currently. However, there are an increasing number of ongoing research and clinical developments on the applications of monoclonal antibodies in the therapy of viral infections without available vaccines, such as Ebola, MERS, and Zika, and infectious diseases without effective anti-viral drugs such as influenza and rabies.[28] The most recent research focuses on the development of monoclonal antibody therapy for treating COVID-19.
References
- ↑ Slifka, Mark K.; Amanna, Ian J. (2018). "Passive Immunization". Plotkin's Vaccines: 84–95.e10. doi:10.1016/B978-0-323-35761-6.00008-0. ISBN 9780323357616. PMC 7151993.
- ↑ "Monoclonal Versus Polyclonal Antibodies: Distinguishing Characteristics, Applications, and Information Resources". ILAR Journal.
- 1 2 "UpToDate". www.uptodate.com. Retrieved 29 March 2022.
- ↑ Coulson, A; Levy, A; Gossell-Williams, M (2014). "Monoclonal Antibodies in Cancer Therapy: Mechanisms, Successes and Limitations". West Indian Medial Journal. 63 (6): 650–654. doi:10.7727/wimj.2013.241. PMC 4663950. PMID 25803383.
- ↑ Strohl, William R.; Strohl, Lila M., eds. (1 January 2012), "8 - Monoclonal antibody targets and mechanisms of action", Therapeutic Antibody Engineering, Woodhead Publishing Series in Biomedicine, Woodhead Publishing, pp. 163–595, doi:10.1533/9781908818096.163, ISBN 978-1-907568-37-4, retrieved 17 April 2022
- ↑ Pelletier, J. Peter R.; Mukhtar, Faisal (1 January 2020), Maitta, Robert W. (ed.), "Chapter 16 - Passive Monoclonal and Polyclonal Antibody Therapies", Immunologic Concepts in Transfusion Medicine, Elsevier: 251–348, doi:10.1016/b978-0-323-67509-3.00016-0, ISBN 978-0-323-67509-3, PMC 7153350
- ↑ "Monoclonal antibody targets and mechanisms of action", Therapeutic Antibody Engineering, Woodhead Publishing Series in Biomedicine, Elsevier, pp. 163–595, 2012, doi:10.1533/9781908818096.163, ISBN 978-1-907568-37-4, retrieved 17 April 2022
- 1 2 Vonderheide, Robert H.; Glennie, Martin J. (1 March 2013). "Agonistic CD40 antibodies and cancer therapy". Clinical Cancer Research. 19 (5): 1035–1043. doi:10.1158/1078-0432.CCR-12-2064. ISSN 1078-0432. PMC 3590838. PMID 23460534.
- ↑ Charles A Janeway, Jr; Travers, Paul; Walport, Mark; Shlomchik, Mark J. (2001). "The complement system and innate immunity". Immunobiology: The Immune System in Health and Disease. 5th Edition.
- ↑ Zeitlin, Larry; Cone, Richard A.; Moench, Thomas R.; Whaley, Kevin J. (1 May 2000). "Preventing infectious disease with passive immunization". Microbes and Infection. 2 (6): 701–708. doi:10.1016/S1286-4579(00)00355-5. ISSN 1286-4579. PMID 10884621.
- ↑ Warwick, Charles A.; Keyes, Alex L.; Woodruff, Trent M.; Usachev, Yuriy M. (1 September 2021). "The complement cascade in the regulation of neuroinflammation, nociceptive sensitization, and pain". Journal of Biological Chemistry. 297 (3): 101085. doi:10.1016/j.jbc.2021.101085. ISSN 0021-9258. PMC 8446806. PMID 34411562.
- ↑ Weiner, Louis M.; Surana, Rishi; Wang, Shangzi (2010). "Monoclonal antibodies: versatile platforms for cancer immunotherapy". Nature Reviews Immunology. 10 (5): 317–327. doi:10.1038/nri2744. ISSN 1474-1733. PMC 3508064. PMID 20414205.
- ↑ Si, Yingnan; Melkonian, Arin L.; Curry, Keegan C.; Xu, Yuanxin; Tidwell, Maranda; Liu, Mingming; Zaky, Ahmed F.; Liu, Xiaoguang (Margaret) (2021). "Monoclonal antibody-based cancer therapies". Chinese Journal of Chemical Engineering. 30: 301–307. doi:10.1016/j.cjche.2020.11.009. S2CID 229393760.
- 1 2 Harding, J.; Burtness, B. (2005). "Cetuximab: An epidermal growth factor receptor chimeric human-murine monoclonal antibody". Drugs of Today. 41 (2): 107–127. doi:10.1358/dot.2005.41.2.882662. ISSN 0025-7656. PMID 15821783.
- 1 2 Kirkpatrick, Peter; Graham, Joanne; Muhsin, Mohamed (July 2004). "Cetuximab". Nature Reviews Drug Discovery. 3 (7): 549–550. doi:10.1038/nrd1445. ISSN 1474-1776. PMID 15272498.
- ↑ "Injection Infusion | ERBITUX (cetuximab)". www.erbitux.com. Retrieved 20 April 2022.
- ↑ Abdulla, Nihal E.; Ninan, Mary J.; Markowitz, Avi B. (April 2012). "Rituximab: Current Status as Therapy for Malignant and Benign Hematologic Disorders". BioDrugs. 26 (2): 71–82. doi:10.2165/11599500-000000000-00000. ISSN 1173-8804. PMID 22339395.
- ↑ Johnson, P W M; Glennie, M J (December 2001). "Rituximab: mechanisms and applications". British Journal of Cancer. 85 (11): 1619–1623. doi:10.1054/bjoc.2001.2127. ISSN 0007-0920. PMC 2363990. PMID 11742477.
- ↑ Weiner, George J. (April 2010). "Rituximab: Mechanism of Action". Seminars in Hematology. 47 (2): 115–123. doi:10.1053/j.seminhematol.2010.01.011. PMC 2848172. PMID 20350658.
- ↑ "Rituximab (Intravenous Route) Side Effects - Mayo Clinic". www.mayoclinic.org. Retrieved 18 April 2022.
- ↑ Connors, Joseph M.; Jurczak, Wojciech; Straus, David J.; Ansell, Stephen M.; Kim, Won S.; Gallamini, Andrea; Younes, Anas; Alekseev, Sergey; Illés, Árpád; Picardi, Marco; Lech-Maranda, Ewa (25 January 2018). "Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin's Lymphoma". New England Journal of Medicine. 378 (4): 331–344. doi:10.1056/NEJMoa1708984. ISSN 0028-4793. PMC 5819601. PMID 29224502.
- ↑ Younes, Anas; Yasothan, Uma; Kirkpatrick, Peter (January 2012). "Brentuximab vedotin". Nature Reviews Drug Discovery. 11 (1): 19–20. doi:10.1038/nrd3629. ISSN 1474-1776. PMID 22212672.
- ↑ "BOOK REVIEWS". Blood. 4 (10): 1182. 1 October 1949. doi:10.1182/blood.v4.10.1182.1182. ISSN 0006-4971.
- 1 2 Fenton, Caroline; Scott, Lesley J; Plosker, Greg L (2004). "Palivizumab: A Review of its Use as Prophylaxis for Serious Respiratory Syncytial Virus Infection". Paediatric Drugs. 6 (3): 177–197. doi:10.2165/00148581-200406030-00004. ISSN 1174-5878. PMID 15170364.
- ↑ "Palivizumab (Intramuscular Route) Side Effects - Mayo Clinic". www.mayoclinic.org. Retrieved 18 April 2022.
- ↑ Jacobson, Jeffrey M.; Kuritzkes, Daniel R.; Godofsky, Eliot; DeJesus, Edwin; Larson, Jeffrey A.; Weinheimer, Steven P.; Lewis, Stanley T. (February 2009). "Safety, Pharmacokinetics, and Antiretroviral Activity of Multiple Doses of Ibalizumab (formerly TNX-355), an Anti-CD4 Monoclonal Antibody, in Human Immunodeficiency Virus Type 1-Infected Adults". Antimicrobial Agents and Chemotherapy. 53 (2): 450–457. doi:10.1128/AAC.00942-08. ISSN 0066-4804. PMC 2630626. PMID 19015347.
- 1 2 Iacob, Simona A.; Iacob, Diana G. (27 November 2017). "Ibalizumab Targeting CD4 Receptors, An Emerging Molecule in HIV Therapy". Frontiers in Microbiology. 8: 2323. doi:10.3389/fmicb.2017.02323. ISSN 1664-302X. PMC 5711820. PMID 29230203.
- ↑ Graham, Barney S.; Ambrosino, Donna M. (May 2015). "History of passive antibody administration for prevention and treatment of infectious diseases". Current Opinion in HIV and AIDS. 10 (3): 129–134. doi:10.1097/COH.0000000000000154. ISSN 1746-630X. PMC 4437582. PMID 25760933.