Ring chromosome 22
| Ring chromosome 22 | |
|---|---|
| Other names: Ring 22 | |
| Symptoms | Intellectual disability, speech delay, hypotonia, hyperactivity |
| Usual onset | Conception |
| Duration | Lifelong |
| Causes | Ring chromosome |
| Diagnostic method | Karyotype |
| Frequency | Lua error in Module:PrevalenceData at line 5: attempt to index field 'wikibase' (a nil value). |
Ring chromosome 22, also known as ring 22, is a rare chromosomal disorder. Ring chromosomes occur when the ends of a chromosome lose material and fuse into a ring shape; in the case of ring 22, this occurs for chromosome 22, the last numbered human autosome. Ring chromosome 22 is marked by a number of consistent traits, such as intellectual disability, speech delay, hypotonia, and hyperactivity. The condition has a similar phenotype to Phelan-McDermid syndrome, as the loss of the SHANK3 gene is implicated in both.
Signs and symptoms
Though ring chromosome 22 has a variable phenotype, a number of consistent features between most cases have been noticed. Most cases have intellectual disability, generally in the moderate to profound range. Other prevalent features include hypotonia (unusual weakness or floppiness of the skeletal muscles), significant hyperactivity, and autism-like features.[1] Significant speech delay is common; in a sample of 35 cases with a median age of 10, over half of the participants were unable to speak, and those who did spoke their first words on average at nearly three years of age.[2] Poor coordination and an unsteady gait are also frequent findings. A number of craniofacial anomalies are common, such as microcephaly, epicanthic folds, and unusually large ears.[3] Other common facial features include almond-shaped eyes with long lashes,[1] thick eyebrows,[4] a bulbous nose, and a "sandal gap" between the toes.[2] Height is usually normal, and physical abnormalities in general mild or absent.[1]
Genitourinary and reproductive abnormalities have been reported. A case is known of an otherwise phenotypically normal man with a ring 22 and azoospermia,[5] and one symptomatic case has been reported of a girl with a malformed clitoris.[2] Though most cases are accompanied by significant disability, reports have been made of intergenerational transmission with some mild or asymptomatic cases. A family has been reported where the ring chromosome 22 was passed down through three generations, with some members demonstrating the traditional symptoms and some not.[6]
Hyperactive behaviour is frequently reported.[7] Bipolar disorder has been reported in adults with ring chromosome 22, and a link between hyperactivity and atypical bipolar disorder is speculated.[8] Autistic-like traits are common in young children, although often abate by preadolescence.[1] Relatively few ring 22 patients fit the full criteria for autism, and it is unclear if such an association genuinely exists or is a consequence of the correlation between autism and developmental delay in general.[9]
Neurofibromatosis type II occurs in a significant minority of cases of ring chromosome 22. The presence of multiple meningiomas is particularly common.[10]
Causes
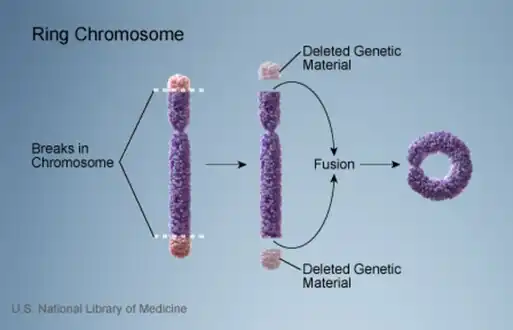
Ring chromosome 22 is caused by a ring chromosome, a form of chromosome mutation where the ends of a chromosome lose genetic material and attach to one another, forming a ring. Chromosome 22 is one of the acrocentric chromosomes, meaning the short arm is unusually small and lacks genes relevant to development; accordingly, the phenotype of ring chromosome 22 is caused only by the loss of genes in the long arm.[1] The amount of genes lost in a deletion can vary significantly, with reports of the loss of anywhere between 0.15% and 21% of chromosome 22 in different symptomatic cases.[2]
Ring chromosomes virtually always arise sporadically, with few documented cases of parent-to-child transmission. Due to the inherent instability of ring chromosomes, inheritance is uncommon even with affected parents.[11] However, cases have been reported of intergenerational inheritance of ring chromosome 22.[6]
Significant overlap exists between the phenotype of ring chromosome 22 and that of Phelan-McDermid syndrome, another chromosome 22 deletion syndrome. This is ascribed to a shared deletion of the SHANK3 gene at 22q13.3. Reports exist of people with ring 22 who lack the SHANK3 deletion and have normal phenotypes, marking SHANK3 as a critical region for the ring chromosome 22 syndrome.[11]
Diagnosis
Ring chromosome 22, like other major chromosomal disorders, is diagnosed via karyotype. Rarely, it may be detected prenatally by amniocentesis or chorionic villus sampling.[3]
Management
As the underlying ring chromosome is an innate genetic disorder, it cannot by itself be treated. Rather, treatment is symptomatic and supportive. Special education is generally indicated due to intellectual disability, while speech therapy may partially overcome speech delays. Physical therapy can assist with hypotonia. Genetic counselling is broadly indicated for potentially heritable genetic disorders.[3]
Epidemiology
Ring chromosome 22 is rare, with around 100 cases reported.[7] It may be more common in females than males,[3] although other researchers have found the opposite.[12]
History
Ring chromosomes 21 and 22 were first identified in 1970. At the time, the two chromosomes could not be differentiated by contemporary cytogenetic analysis and were both referred to as the "G chromosome", with ring chromosome 22 being referred to as "G-deletion syndrome II"; as techniques advanced, G-deletion syndrome II was identified as ring chromosome 22.[13] Early reports were split on the topic of whether ring chromosome 22 constituted a consistent syndrome or simply a finding in a heterogeneous group of intellectually disabled people.[14] One early case that drew attention to the syndrome was a report of monozygotic twin sisters with a ring 22, one of the first recorded reports of a shared chromosomal anomaly between twins.[15]
See also
References
- 1 2 3 4 5 Unique, Jeffries A, Hultén M (2014). "Ring 22" (PDF). Unique. Archived (PDF) from the original on 21 March 2021. Retrieved 16 March 2021.
- 1 2 3 4 Jeffries AR, Curran S, Elmslie F, Sharma A, Wenger S, Hummel M, Powell J (29 July 2005). "Molecular and phenotypic characterization of ring chromosome 22". American Journal of Medical Genetics. 137A (2): 139–147. doi:10.1002/ajmg.a.30780. PMID 16059935. S2CID 21211829.
- 1 2 3 4 Kulkarni S (2009). "Chromosome 22 Ring". National Organization for Rare Disorders. Archived from the original on 5 May 2021. Retrieved 16 March 2021.
- ↑ Ishmael HA, Cataldi D, Begleiter ML, Pasztor LM, Dasouki MJ, Butler MG (7 May 2003). "Five new subjects with ring chromosome 22". Clinical Genetics. 63 (5): 410–414. doi:10.1034/j.1399-0004.2003.00064.x. PMC 6714054. PMID 12752574.
- ↑ Zuccarello D, Dallapiccola B, Novelli A, Foresta C (2010). "Azoospermia in a man with a constitutional ring 22 chromosome". European Journal of Medical Genetics. 53 (6): 389–391. doi:10.1016/j.ejmg.2010.07.014. PMID 20709628.
- 1 2 Stoll C, Roth MP (May 1983). "Segregation of a 22 ring chromosome in three generations". Human Genetics. 63 (3): 294–296. doi:10.1007/BF00284669. PMID 6852827. S2CID 33812679.
- 1 2 "Ring chromosome 22". Genetic and Rare Disease Information Center. 1 February 2021. Archived from the original on 18 March 2021. Retrieved 16 March 2021.
- ↑ Sovner R, Stone A, Fox C (February 1996). "Ring chromosome 22 and mood disorders". Journal of Intellectual Disability Research. 40 (1): 82–86. doi:10.1111/j.1365-2788.1996.tb00607.x. PMID 8930062.
- ↑ MacLean JE, Teshima IE, Szatmari P, Nowaczyk MJM (23 March 2000). "Ring chromosome 22 and autism: Report and review". American Journal of Medical Genetics. 90 (5): 382–385. doi:10.1002/(SICI)1096-8628(20000228)90:5<382::AID-AJMG7>3.0.CO;2-T. PMID 10706359.
- ↑ Zirn B, Arning L, Bartels I, Shoukier M, Hoffjan S, Neubauer B, Hahn A (20 November 2010). "Ring chromosome 22 and neurofibromatosis type II: proof of two‐hit model for the loss of the NF2 gene in the development of meningioma". Clinical Genetics. 81 (1): 82–87. doi:10.1111/j.1399-0004.2010.01598.x. PMID 21175598. S2CID 20944316.
- 1 2 Yip MY (2015). "Autosomal ring chromosomes in human genetic disorders". Translational Pediatrics. 4 (2): 164–174. doi:10.3978/j.issn.2224-4336.2015.03.04. PMC 4729093. PMID 26835370.
- ↑ Mahajan S, Kaur A, Singh JR (2012). "Ring Chromosome 22: A Review of the Literature and First Report from India". Balkan Journal of Medical Genetics. 15 (1): 55–59. doi:10.2478/v10034-012-0009-8. PMC 3776658. PMID 24052724.
- ↑ Warren RJ, Rimoin DL, Summitt RL (1973). "Identification by fluorescent microscopy of the abnormal chromosomes associated with the G-deletion syndromes". American Journal of Human Genetics. 25 (1): 77–81. PMC 1762232. PMID 4265215.
- ↑ Hunter AGW, Ray M, Wang HS, Thompson DR (October 1977). "Phenotypic correlations in patients with ring chromosome 22". Clinical Genetics. 12 (4): 239–249. doi:10.1111/j.1399-0004.1977.tb00933.x. PMID 912941. S2CID 40631541.
- ↑ Lindenbaum RH, Bobrow M, Barber L (March 1973). "Monozygotic Twins with Ring Chromosome 22". Journal of Medical Genetics. 10 (1): 85–89. doi:10.1136/jmg.10.1.85. PMC 1012981. PMID 4697858.