Sanjad-Sakati syndrome
| Sanjad-Sakati syndrome (middle east syndrome ) | |
|---|---|
| Other names: Hypoparathyroidism-short stature-intellectual disability-seizures syndrome | |
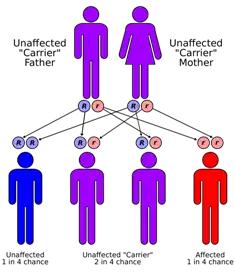 | |
| Sanjad-Sakati syndrome is inherited in an autosomal recessive manner | |
| Usual onset | diagnosis is clinically by features of triad of growth retardation, dysmorphic features and hypo calcemic tetany or sezures |
Sanjad-Sakati syndrome ( middle east syndrome )is a rare autosomal recessive genetic condition seen in offspring of Middle Eastern origin. It was first described in Saudi Arabia,[1] but has been seen in Qatari, Kuwaiti, Omani and other children from the Middle East as well as elsewhere.[2] The condition is caused by mutations or deletions in the TBCE gene of Chromosome No.1.
The condition is characterised by a triad of growth retardation and intellectual disability, hypoparathyroidism and dysmorphism.
Symptoms and signs
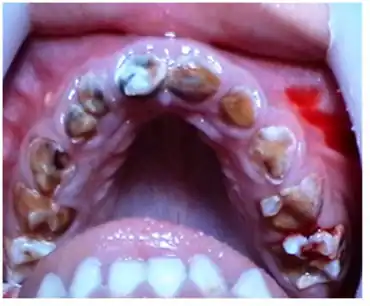
Children with the Sanjad Sakati syndrome have a triad of hypoparathyroidism (with episodes of hypocalcemia, hypocalcemic tetany and hypocalcemic seizures), severe intellectual disability and dysmorphism. Typically, children with this syndrome are born low-birth-weight due to intrauterine growth retardation. At birth, there is dysmorphism, which is later typified into the features described below. The child is stunted, often with demonstrable growth hormone deficiency and has moderate to severe intellectual disability, mainly as a consequence of repeated seizures brought on by the low blood ionic calcium levels. The immuno-reactive parathormone levels are low to undetectable, with low calcium and high phosphate levels in the blood.
Dysmorphism is most evident on the face, with the following features:
- Long narrow face
- Deep-set, small eyes
- Beaked nose
- Large, floppy ears
- Small head (microcephaly) and
- Thin lips with a long philtrum.
Other features
Other features include:
- Stunting
- Small hands and feet with long, tapering fingers and clinodactyly
- Dental anomalies in the form of malalignment and malocclusion
In another study of six patients, there were low levels of IGF-1 and markedly retarded bone age.[3]
Genetics
This disorder is caused by an abnormality of the TBCE gene,[4] the locus for which is on Chromosome 1q42.3. The locus is a 230 kb region of gene with identified deletions and mutations in affected individuals.[5] There are rare cases of the disorder not being due to a TBCE gene abnormality.[6]
Diagnosis
The diagnosis of this condition, Sanjad-Sakati syndrome, can be done via:[7]
- Medical exam
- Lab test
- Genetic test
- Physical presentation
Management
Management is mainly supportive by controlling seizures and blood calcium levels.
History
First reported from Saudi Arabia in 1988, Sanjad-Sakati syndrome,[8] also known as Hypoparathyroidism-Retardation-Dysmorphism (HRD) syndrome, or less commonly as the Middle East syndrome, is a very rare genetically inherited disorder seen in the Middle East and children of Middle Eastern origin elsewhere in the world. The condition is named after Sami A. Sanjad and Nadia Awni Sakati.
References
- ↑ Sanjad, S (1988). "Congenital hypoparathyroidism with dysmorphic features: a new syndrome. (Abstract)". Pediatric Research. 23: 271A.
- ↑ Sanjad, S. A.; Sakati, N. A.; Abu-Osba, Y. K.; Kaddoura, R.; Milner, R. D. (February 1991). "A new syndrome of congenital hypoparathyroidism, severe growth failure, and dysmorphic features". Archives of Disease in Childhood. 66 (2): 193–196. doi:10.1136/adc.66.2.193. ISSN 1468-2044. PMC 1792808. PMID 2001103.
- ↑ Hershkovitz, E.; Shalitin, S.; Levy, J.; Leiberman, E.; Weinshtock, A.; Varsano, I.; Gorodischer, R. (May 1995). "The new syndrome of congenital hypoparathyroidism associated with dysmorphism, growth retardation, and developmental delay--a report of six patients". Israel Journal of Medical Sciences. 31 (5): 293–297. ISSN 0021-2180. PMID 7538982.
- ↑ "OMIM Entry - * 604934 - Tubulin-Specific Chaperone E; TBCE". Omim.org. Archived from the original on 2015-09-27. Retrieved 2015-08-25.
- ↑ Parvari, R., Hershkovitz, E., Grossman, N., Gorodischer, R., Loeys, B., Zecic, A., Mortier, G., Gregory, S., Sharony, R., Kambouris, M., Sakati, N., Meyer, B. F., and 10 others. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nature Genet. 32: 448-452, 2002.
- ↑ Courtens, W., Wuyts, W., Poot, M., Szuhai, K., Wauters, J., Reyniers, E., Eleveld, M., Diaz, G., Nothen, M. M., Parvari, R. Hypoparathyroidism-retardation-dysmorphism syndrome in a girl: a new variant not caused by a TBCE mutation--clinical report and review. Am. J. Med. Genet. 140A: 611-617, 2006.
- ↑ "Hypoparathyroidism-intellectual disability-dysmorphism syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 24 September 2021. Retrieved 23 September 2021.
- ↑ "OMIM Entry - # 241410 - Hypoparathyroidism-Retardation-Dysmorphism Syndrome; HRD". Omim.org. Archived from the original on 2015-09-24. Retrieved 2015-08-25.
External links
| Classification | |
|---|---|
| External resources |
|