T-Cell Acute Lymphoblastic Leukemia
| T-Cell Acute Lymphoblastic Leukemia (T-ALL) | |
|---|---|
| Specialty | Haematology, oncology |
| Symptoms | Recurrent infections, unusual or common bleeding and bruising, extreme tiredness, unexplained fever, unexplained weight gain, swollen lymph nodes |
| Usual onset | Most prevalent in the adult population with incidences diminishing with age. Amongst pediatric population, median onset of age 9. Marked male predominance [1] |
| Causes | Currently unknown |
| Diagnostic method | Blood test, bone marrow aspiration,[2] biopsy, CT, MRI, lumbar puncture,[2] genetic testing |
| Treatment | Long-term chemotherapy,[3] CNS radiation therapy,[1] stem cell transplantation [4] |
| Prognosis | 5-Year Event Free Survival: 70%, Overall Survival: 80% [1] |
| Frequency | 7% at ages 1-10, 14% at ages 10-15, and 29% at ages 15-18 [5] |
T-Cell Lymphoblastic Leukemia (T-ALL) is a type of acute lymphoblastic leukemia with aggressive malignant neoplasm of the bone marrow.[6] Acute Lymphoblastic Leukemia (ALL) is a condition where immature white blood cells accumulate in the bone marrow, subsequently crowding out normal white blood cells[7] and create build-up in the liver, spleen, and lymph nodes.[8] The two most common types of ALL are B-Lymphocytes and T-Lymphocytes, where the first protects the body against viruses and bacteria through antibody production which can directly destroy target cells or trigger others to do so, whilst the latter directly destroy bacteria or cells infected with viruses.[9] Approximately 20% of all ALL patients are categorized specifically to suffer from T-ALL and it is seen to be more prevalent in the adult population in comparison to children, with incidences shown to diminish with age.[6][10] Amongst T-ALL cases in the pediatric population, a median onset of age 9 has been identified and the disease is particularly prominent amongst adolescents.[6] The disease stems from cytogenic and molecular abnormalities, resulting in disruption of developmental pathways controlling thymocyte development, tumor suppressor development, and alterations in control of cell growth and proliferation.[1] Distinct from adult T-Cell Leukemia where T-Cell Lymphotropic Virus Type I causes malignant maturation of T-cells, T-ALL is a precursor for lymphoid neoplasm.[6] Its clinical presentation most commonly includes infiltration of the central nervous system (CNS), and further identifies mediastinal mass presence originating from the thymus, along with extramedullary involvement of multiple organs including the lymph node as a result of hyperleukocytosis.
Signs and symptoms
T-ALL patients may not always experience the all signs and symptoms below. Patients with other medical conditions that are not leukemia may also experience similar symptoms.
- Recurrent infections due to lack of normal white blood cells (neutrophils)[11]
- Unusual and/or common bleeding and bruising
- Extreme tiredness and swellings in the neck (lymph nodes) or the middle of the chest, causing possible facial swelling
- Unexplained fevers, chills, and/or night sweats
- Unexplained weight loss and/or loss of appetite
- Swollen lymph nodes
- Unexplained skin itch
Clinical manifestations
Originating from epigenetic and genetic alterations in immature thymocytes, T-ALL is a highly aggressive and heterogenous disease. Patients often present extensive bone marrow involvement, mediastinal mass, adenopathy, CNS involvement, and splenomegaly.[1] Symptoms can be presented acutely or develop progressively over time. The most common clinical feature amongst patients is the proliferation of malignant clones, hence suppressing normal hematopoiesis, resulting in deficiency of functioning peripheral blood cells (particularly thrombocytes) deficiency.[1]
Risk factors
T-ALL is not a contagious nor inherited condition. Its two main risk factors are age and gender.[8] Most cases of leukemia increase with age, with ALL being the main exception, which peaks in children aged 2 to 5 years. T-ALL is seen to be most prevalent in the adult population, but amongst cases in the pediatric population, it is seen to have a median onset of age 9 and is most prominent to adolescents.[6][10] The disease also is marked male predominance with a three-fold increased risk of developing T-ALL in comparison to females. It is currently unclear as to why T-ALL is preferential towards older children and males.[1]
Cytogenetics
Basic karyotyping showed structural chromosomal rearrangements in 50-75% of T-ALL patients, primarily inversion and translocations.[1] Diagnostic yield can be substantially increased through further diagnosis through Fluorescent In Situ Hybridization (FISH) and other various molecular technologies, for example Single Nucleotide Polymorphism (SNP) array. The most common structural abnormality is rearrangement of the TCR gene. 95% of T-Cell TCR consist of an alpha and beta chain (encoded by TRA and TRB, respectively), where only 5% of T-Cell TCR consists of gamma and delta chains (encoded by TRG and TRD, respectively).[4]
Karyotyping showed that TRD and TRB undergo recombination most commonly, whereas TRA is seldom involved and TRG is rarely rearranged. These rearrangements affect the normal process of TCR and could lead to cellular machinery failing to correctly repair recombination-activating RAG protein induced double-strand breaks (DSBs).[1] All 30 genes known to illegitimately recombine with TCR genes function primarily to regulate epigenetics through roles such as signal transducers, transcription factors (tumor suppressors or oncogenes), cell cycle regulators, or ribosomal proteins.
T-Cell TCR encoded by TRA, TRD, and TRG at chromosome bands 14q11 and 7q34 become malignant in T-ALL patients.[1] The build-up of malignant T-cells in T-ALL are clones with identical T-cell receptor gene arrangements having taken rise from a single cell. The gene rearrangements, as a result of the malignant cell, juxtapose both TCR genes and other critical genes that code for transcription factors. This results in dysregulation of partner gene transcription, which serves as the main cause of leukemogenesis - a multi-step process of induction, development, and progression of leukemic diseases.[1] 20% of all leukemias demonstrate simultaneous rearrangement of these genes.
Pathology
Like most cancers, mutations in the DNA begin T-ALL development and lead to loss of function of white blood cells. Different subtypes of leukemia have similarities in their causes, which are a combination of genetics, epigenetic changes, and environmental factors. However, because there are few T-ALL cases in comparison to other subtypes of leukemia, there is currently no clear cause of T-ALL. T-ALL is not contagious nor inherited but specific genetic mutations, commonly including NOTCH1 and CDKN2A, may be passed along which increases susceptibility of T-ALL.[10]
Causes of T-ALL
Genetic conditions
Some patients may have familial histories with leukemia predispositions which increases risk of developing T-ALL. Li-Fraumeni syndrome is an inherited condition that leads to mutation of TP53, a tumor suppressor gene, which then increases risk of T-ALL. Mutation in gene SPRED1 is also associated with development of T-ALL.
Patients with immature thymocytes in the thymus begins T-ALL development. Furthermore, hereditary conditions such as Down syndrome, neurofibromatosis type 1, ataxia telangiectasia, and Noonan syndrome are associated with higher risk of developing T-ALL.
Radiation exposure
Those who have had pervious chemotherapy and exposure to radiation may have increased risks of developing T-ALL. CDKN2A is an inherited polymorphism variant that is seen to be associated with development of T-ALL. SR-90 emission from nuclear reactor accidents is also believed to increase risk of developing T-ALL.
Chemical exposure
Benzene, a chemical classified as being carcinogenic to humans, is associated with increased risk of T-ALL, as well as other forms of leukemia.[12]
Viruses
Human T-Lymphotropic virus (HTLV-1) is a retroviral infection that affect white blood cells (T cells), which may later develop into T-ALL and other subtypes of leukemia.[13]
Diagnosis
When doctors are suspicious of a patient potentially suffering from T-ALL after careful examination of background (including medical history, signs, and symptoms), doctors would then conduct tests, procedures, and scans to proceed with diagnosis of T-ALL. Some symptoms and medical history may not be specific enough to diagnose T-ALL, so further testing may be required. Doctors may consider some factors mentioned but would not necessarily conduct all tests possible.[11]
Assessments
Blood tests
Complete blood count (CBC) is done to test for T-ALL by measuring the different types and maturity of cells in the patient's blood, which allows the donor to determine whether leukemic cells are present in the patient. Additionally, blood tests that show high levels of white blood cells or low levels of red blood cells may also be a sign of T-ALL. Further testing could also help indicate whether T-ALL has affected other organs such as the kidneys as well as the genetic alterations of the disease.
Bone marrow aspiration and biopsy
Bone marrow consists of a combination of solid and liquid components. Bone marrow aspiration and biopsies are typically done simultaneously to help determine and confirm the type and severity of T-ALL. Further biopsies such as skin and lymph node biopsies may also need to be done to check for the spread of T-ALL.[2]
X-rays and ultrasound
As swollen spleen and lymph nodes are symptoms of T-ALL, X-rays and ultrasound scans, such as CT and MRI, can help confirm the diagnosis. This also provides information on the impact T-ALL has on other organs of the body.
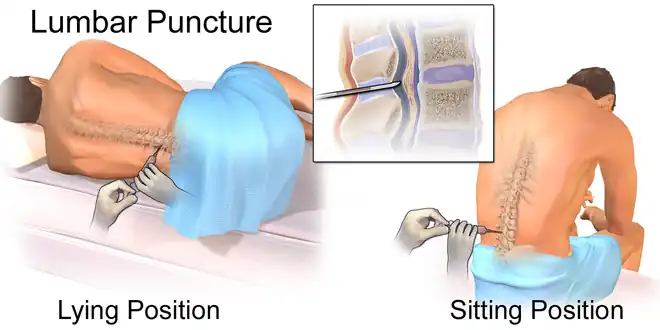
Lumbar puncture
To prevent ineffective treatments towards T-Cells that have invaded the CNS, lumbar puncture allows doctors to determine whether the treatments will be effective. This also reveals the spread of T-ALL.[2]
Genetic test
Genetic testing helps identify chromosomal abnormalities in the patients. This can help identify the genetic mutations and therefore diagnose the specific leukemia subtype.
Staging
Normal staging is not used for T-ALL because it is already spread throughout the body when first diagnosed. However, they have their own system of classifying T-ALL cases.[14] First, patterns of gene expression are investigated to define T-ALL. Then, stages of thymic development can be determined by identifying specific expressions in chromosomal abnormalities. This forms the stages of T-ALL cases being either at high or low risk.[8] Patients will then receive the appropriate treatment in respect to whichever class they are in.[14]
Treatment
Currently, standard treatment of T-ALL takes the form of long-term chemotherapy and drug intake to prevent or treat side effects associated with low white blood cell count as a result of intensive chemotherapy regimes. The treatment typically takes place over three stages: induction, consolidation, and maintenance.[3] Treatment is expected to span over approximately two years with the maintenance phase lasting the longest. T-ALL can spread to areas of the brain and spinal cord,[2] which can be diagnosed through lumbar puncture assessment in patients suspected to suffer from T-ALL. Lumbar puncture helps to identify leukemic cells surrounding the cerebrospinal fluid (CSF).[3] Even if leukemic cells are not found in the CSF at the time of diagnosis, it is highly likely that they will spread there with time and progression of the disease. Henceforth, Prophylactic Intrathecal Chemotherapy in CNS lymphoma, a treatment to lower risk of leukemia spreading to the spinal cord and brain by directly administering chemotherapy to the CSF, is crucial.[3]
In comparison to B-ALL, T-ALL patients present more high-risk features including tendency for earlier relapse, CNS involvement, and resistance to chemotherapy. In response, Prophylactic Intrathecal Chemotherapy is further enhanced with CNS radiation therapy.[1] In treating high-risk T-ALL patients, allogeneic hematopoietic stem cell transplantation has been deemed to produce highly successful and promising results. However, its consequence includes increased relapse, which reduces its curative potential. Patients undergoing transplantation must be continuously monitored for Minimal Residual Disease (MRD), usually via qPCR analysis of T-Cell Receptor (TCR) genes to evaluate for fusion transcripts such as SIL-TAL1.[15] Mutation of TAL1 is frequently present in T-ALL patients, where SIL/TAL1 fusion gives rise to inappropriate TAL1 expression, in turn promoting T-Cell leukemogenesis.[4] The analysis is critical to ensure that immediate intervention is taken during early stages of relapse.
Young T-ALL patients showed significant improvement through multimodal therapy, involving initial induction therapy – including a glucocorticoid, vincristine, L-asparaginase, and an anthracycline - for 4 to 6 weeks, intensive combination therapy for 6–8 months, lastly 18–30 months of low-intensity anti-metabolite-based therapy.[8] It is crucial to note the importance of differential treatment amongst youth and adults. Studies have shown that through administering a random variation of either traditional pediatric scheme or intensive block-based chemotherapy, the two groups showed significantly different responses.[4] Although both treatments included administering high-dose methotrexate and asparaginase and allogeneic hematopoietic stem cell transplantation, high survival and low death rates were present for all patients for the first treatment whereas the latter led to a high toxic death rate amongst adults.[16]
Prognosis
In childhood, T-ALL patients can expect a 5-year event free survival and overall survival of, respectively, 70% and 80%.[1] Amongst approximately 25% of children who relapse, survival rate sits at 30-50% and the patients show much poorer prognosis.[1] Monitoring for MRD is critical as previously mentioned, through qPCR analysis, in order to evaluate the efficacy of treatment.
Recent genomic studies have found that a selection of genetic variants in relation to clonal evolution that drive resistance have been found as the basis for T-ALL relapse. Over 20% of patients with relapsed T-ALL showed mutation in the variant cytosolic 5’-nucleotidase II (NT5C2) gene, while the TFDP3 gene has also been found to confer chemoresistance in children.[1]
Epidemiology
Although over 100 genes mutations have been identified in T-ALL patients, only NOTCH1 and CDKN2A mutations are considered to be common.[13]
In over 50% of pediatric T-ALL cases, mutations in epigenetic regulators have been identified.[5] This activates mutations of NOTCH1 and gene FBXW7 causes the tumor-suppressing gene to lose its functions, leading to T-ALL.[17]
Near-telomeric location may sometimes generate subtle exchanges in DNA material at the loci involved in oncogenic rearrangements of T-ALL. This causes cryptic translocation and therefore deletes the putative tumor suppressor gene CDKN2A (INK4A). At the same time, TLX1 and NOTCH1 may also be activated at higher frequency than usual. The multistep prognosis of T-ALL has hence been said to intensify and rapidly progress due to accumulation of effects resulting from dysregulation of multiple signaling pathways.[5]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 "Pediatric T-Cell Acute Lymphoblastic Leukemia". atlasgeneticsoncology.org. Retrieved 2020-04-07.
- 1 2 3 4 5 "Acute lymphoblastic leukemia (ALL): Symptoms, causes, and treatment". www.medicalnewstoday.com. 14 October 2019. Retrieved 2020-04-07.
- 1 2 3 4 "Typical Treatment of Acute Lymphocytic Leukemia (ALL)". www.cancer.org. Retrieved 2020-04-07.
- 1 2 3 4 D’Angiò, Mariella; Valsecchi, Maria G.; Testi, Anna M.; Conter, Valentino; Nunes, Vittorio; Parasole, Rosanna; Colombini, Antonella; Santoro, Nicola; Varotto, Stefania; Caniglia, Maurizio; Silvestri, Daniela (January 2015). "Clinical features and outcome of SIL/TAL1-positive T-cell acute lymphoblastic leukemia in children and adolescents: a 10-year experience of the AIEOP group". Haematologica. 100 (1): e10–e13. doi:10.3324/haematol.2014.112151. ISSN 0390-6078. PMC 4281327. PMID 25304610.
- 1 2 3 "T-lineage acute lymphoblastic leukemia (T-ALL)". atlasgeneticsoncology.org. Retrieved 2020-04-07.
- 1 2 3 4 5 Litzow, Mark R.; Ferrando, Adolfo A. (2015-08-13). "How I treat T-cell acute lymphoblastic leukemia in adults". Blood. 126 (7): 833–841. doi:10.1182/blood-2014-10-551895. ISSN 0006-4971. PMID 25966987.
- ↑ "Acute Lymphoblastic Leukemia (ALL)". www.stjude.org. Retrieved 2020-04-07.
- 1 2 3 4 "T-cell Acute Lymphoblastic Leukaemia". Leukaemia Care. Retrieved 2020-04-07.
- ↑ "What is the Difference Between B-cell Lymphoma and T-cell Lymphoma?". Dana-Farber Cancer Institute. 18 Jun 2019. Retrieved 7 Apr 2020.
- 1 2 3 "T-Cell Acute Lymphoblastic Leukemia - My Cancer Genome". www.mycancergenome.org. Retrieved 2020-04-07.
- 1 2 "Leukemia - Chronic T-Cell Lymphocytic - Stages". Cancer.Net. 2012-06-25. Retrieved 2020-04-07.
- ↑ Khalade, Abdul; Jaakkola, Maritta S.; Pukkala, Eero; Jaakkola, Jouni J. K. (2010-06-28). "Exposure to benzene at work and the risk of leukemia: a systematic review and meta-analysis". Environmental Health: A Global Access Science Source. 9: 31. doi:10.1186/1476-069X-9-31. ISSN 1476-069X. PMC 2903550. PMID 20584305.
- 1 2 "Human T-cell leukemia virus type 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2020-04-07.
- 1 2 "Acute lymphocytic leukemia - Diagnosis and treatment - Mayo Clinic". www.mayoclinic.org. Retrieved 2020-04-07.
- ↑ D’Angiò, Mariella; Valsecchi, Maria G.; Testi, Anna M.; Conter, Valentino; Nunes, Vittorio; Parasole, Rosanna; Colombini, Antonella; Santoro, Nicola; Varotto, Stefania; Caniglia, Maurizio; Silvestri, Daniela (2015-01-16). "Clinical features and outcome of SIL/TAL1-positive T-cell acute lymphoblastic leukemia in children and adolescents: a 10-year experience of the AIEOP group". Haematologica. 100 (1): e10–e13. doi:10.3324/haematol.2014.112151. ISSN 0390-6078. PMC 4281327. PMID 25304610.
- ↑ Quist-Paulsen, P.; Toft, N.; Heyman, M.; Abrahamsson, J.; Griškevičius, L.; Hallböök, H.; Jónsson, Ó G.; Palk, K.; Vaitkeviciene, G.; Vettenranta, K.; Åsberg, A. (2020-02-20). "T-cell acute lymphoblastic leukemia in patients 1–45 years treated with the pediatric NOPHO ALL2008 protocol". Leukemia. 34 (2): 347–357. doi:10.1038/s41375-019-0598-2. ISSN 1476-5551. PMID 31611626. S2CID 204459614.
- ↑ Belver, Laura; Ferrando, Adolfo (2016-08-30). "The genetics and mechanisms of T cell acute lymphoblastic leukaemia". Nature Reviews Cancer. 16 (8): 494–507. doi:10.1038/nrc.2016.63. ISSN 1474-1768. PMID 27451956. S2CID 28636912. Retrieved 2020-04-05.