Pallister–Killian syndrome
| Pallister-Killian syndrome | |
|---|---|
| Other names: Tetrasomy 12p mosaicism, Pallister mosaic aneuploidy syndrome | |
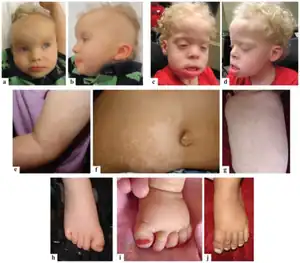 | |
| (a-d) Two children with PKS (e-g) Decreased and increased pigmentation of skin (h-j) Broad toes | |
| Specialty | Medical genetics, pediatrics |
| Symptoms | Multiple birth defects |
| Usual onset | Prenatally |
| Causes | Small supernumerary marker chromosome |
The Pallister–Killian syndrome (PKS), also termed tetrasomy 12p mosaicism or the Pallister mosaic aneuploidy syndrome, is an extremely rare and severe genetic disorder. PKS is due to the presence of an extra and abnormal chromosome termed a small supernumerary marker chromosome (sSMC). sSMCs contain copies of genetic material from parts of virtually any other chromosome and, depending on the genetic material they carry, can cause various genetic disorders and neoplasms. The sSMC in PKS consists of multiple copies of the short (i.e. "p") arm of chromosome 12.[1] Consequently, the multiple copies of the genetic material in the sSMC plus the two copies of this genetic material in the two normal chromosome 12's are overexpressed and thereby cause the syndrome.[2] Due to a form of genetic mosaicism, however, individuals with PKS differ in the tissue distributions of their sSMC and therefore show different syndrome-related birth defects and disease severities. For example, individuals with the sSMC in their heart tissue are likely to have cardiac structural abnormalities while those without this sSMC localization have a structurally normal heart.[3]
PKS was first described by Philip Pallister in 1977 and further researched by Maria Teschler-Nicola and Wolfgang Killian in 1981.[4]
Signs and symptoms
Individuals with PKS present prenatally or at birth with multiple birth defects. These defects include: brain atrophy, agenesis of the corpus callosum, polymicrogyria of the brain, and/or spot calcifications in the brain's lateral sulcus; deafness and/or blindness; autonomic nervous system dysfunctions such as anhidrosis, hypohidrosis, and/or episodic spells of hyperventilation interspersed with breath-holding; symptoms of spinal cord malformations; profound or less commonly mild to severe intellectual disability; epileptic seizures; heart and/or anal defects; diaphragmatic hernias; marked muscle weakness; supernumerary nipples; abnormal facial features such as frontal bossing, high frontal hairline, balding around the temple and frontal areas, sparse eyebrows and lashes, hypertelorism, small and flat nose, full cheeks, long philtrum, large mouth with downturned corners, thin cupid's bow-shaped upper lip, micrognathia (i.e. undersized jaw), disformed ears that are low-set, thick eyebrows, and/or prominent lips and chin; abnormal oral/dental features such as enlarged tongue, overgrowth of the alveolar ridge and/or gums, delayed teeth eruption, and/or missing or double teeth; patchy skin depigmentations; skeletal anomalies such as limb shortening, lymphedema, increased soft tissues in the extremities, short/broad palms and/or fingers, and/or clinodactyly of the fifth fingers or toes; excessive prenatal and birth weights followed by postnatal declines in growth rates; delayed closure of the anterior fontanel; and/or delayed puberty in males but nor females.[3]
Causes
PKS is caused by an sSMC that consists of two copies or, less commonly, four copies of the genetic material in the p arm of chromosome 12.[1][3] Recent studies in two individuals with PKS found their sSMCs consisted specifically of genetic material located in a stretch of chromosome 12 's p arm starting at its band 11 and running to its end. This area, called the PKS critical region, contains three genes, ING4, CHD4, and MFAP5 (also termed the MAGP2 gene), which are candidates for contributing to the development of the syndrome.[3]
One suggested mechanism for the development of the sSMC in PKS involves three sequential events: 1) chromosome 12 suffers a nondisjunction, i.e. a failure of its homologous chromosomes or sister chromatids to separate properly during the second meiosis cell division that forms maternal eggs; 2) while most of the eggs suffering this nondisjunction die, a rare egg with the nondisjunction acquires a second structural aberration, isochromosome formation, that results in the creation of an extra chromosome consisting of copies of two or four p arms but no q arms of chromosome 12, i.e. the sSMC; and 3) the sSMC-contianing egg, after being fertilized by a genetically normal sperm, develops into an offspring containing copies of this sSMC in some but not all cells, tissues, and/or organs consequently have some but not all of the defects associated with PKS. This mechanism applies only to female parents who are by far the most common originators of the sSMC in PKS. The mechanism explaining the few cases in which male parents form a sperm containing this sSMC has not yet been clearly formulated.[2]
Diagnosis
Before birth
PKS is commonly diagnosed by detecting its causative sSMC as defined by identifying the overexpression of its genetic material. This method has detected the sSMC and therefore diagnosed a fetus as having PKS based on genomic analyses of fetal skin fibroblasts, placenta chorionic villi, cells isolated from the amniotic fluid,[4] fibroblasts isolated from the fetus's umbilical cord,[5] and cells isolated from the fetus's umbilical cord blood.[6] PKS can also be diagnose using fetal ultasound imaging methods. Ultrasound imaging in PKS commonly find fetuses that are too large for their gestational age, contain polyhydramnios (excess amniotic fluid in their amniotic sacs, and have rhizomelic limbs (shortening of the proximal part of the limbs). Less commonly, the imaging evidences diaphragmatic hernias and/or various other major PKS structural malformations. In most cases, however, the ultrasound findings are not diagnostic of PKS. Furthermore, ultrasound diagnoses is best applied in the second or third pregnancy trimester when structural anomalies are more clearly defined and detectable.[7]
Because the prenatal diagnosis of PKS using the methods just cited is difficult, often indecisive, and/or best employed later in a woman's pregnancy, prenatal cell-free DNA screening (cfDNA screening), also known as noninvasive prenatal screening, has been used to diagnose PKS. This method can diagnose PKS in 10 week and older fetuses. In cfDNA screening, DNA from a mothers blood is extracted and screened for the presence of specific chromosome abnormalities such as those associated with the Down syndrome, Patau syndrome (also termed trisomy 13[8]), and Edwards syndrome (also termed trisomy 18[9]). (Small amounts of a fetus's DNA escapes through the placenta to circulate in the mothers blood.[10]) A genome-wide association study[11] done in China used genome-wide cfDNA analyses to diagnose various chromosome-related disorders including PKS. The study scanned the DNA in the blood of 29,007 pregnant women and found three cases with abnormal amount of DNA originating from the entire p-arm of chromosome 12. All three cases were confirmed to have a fetus with PKS. Two of these cases were missed by chromosomal microarray analysis of the placenta and chorionic villi. However, the study did not define the rate of false negative cases, i.e. negative results in women actually carrying a fetus with PKS. While further studies are required, this method may turn out to be a critically useful addition for the prenatal detection of PKS, particularly during early pregnancies for the purpose of pregnancy options counseling.[12]
After birth
The postnatal diagnosis of PKS is strongly suggested or indicated in most cases based on finding the key defects of PKS in an individual on physical examination and various radiography, ultrasound, and related methods.[3] However, some individuals with this syndrome do not have a sufficient number of these defects or have a set of defects that are also compatible with other birth defect disorders such as Fryns syndrome, trisomy 12p,[13] and Sifrim-Hitz-Weiss syndrome (also termed CHD4 Neurodevelopmental Disorder[14]).[3] The diagnosis can be confirmed in these cases as well as in all cases of PKS by detecting its sSMC using special methods. This sSMC has been successfully detected (>90% of confirmed cases) in the DNA extracted by a buccal swab taken from the inside of an individual's cheek or the DNA extracted form an individual's cultured skin fibroblasts, i.e. fibroblasts from a skin biopsy grown in a laboratory for at least several days. The sSMC in these tissues or cells is identified by multiplex ligation-dependent probe amplification (i.e. MLPA)[3] or microarray-based comparative genomic hybridization (i.e. array CGH).[2] Because of mosaicism, testing an individual's circulating blood lymphocytes only rarely detects (i.e. gives mostly false negative results) in true PKS cases.[3][15][16]
Management
There is no treatment, however children could benefit from special education.[17]
See also
References
- 1 2 Jafari-Ghahfarokhi H, Moradi-Chaleshtori M, Liehr T, Hashemzadeh-Chaleshtori M, Teimori H, Ghasemi-Dehkordi P (2015). "Small supernumerary marker chromosomes and their correlation with specific syndromes". Advanced Biomedical Research. 4: 140. doi:10.4103/2277-9175.161542. PMC 4544121. PMID 26322288.
- 1 2 3 Karaman B, Kayserili H, Ghanbari A, Uyguner ZO, Toksoy G, Altunoglu U, Basaran S (2018). "Pallister-Killian syndrome: clinical, cytogenetic and molecular findings in 15 cases". Molecular Cytogenetics. 11: 45. doi:10.1186/s13039-018-0395-z. PMC 6098576. PMID 30140312.
- 1 2 3 4 5 6 7 8 Arghir A, Popescu R, Resmerita I, Budisteanu M, Butnariu LI, Gorduza EV, Gramescu M, Panzaru MC, Papuc SM, Sireteanu A, Tutulan-Cunita A, Rusu C (May 2021). "Pallister-Killian Syndrome versus Trisomy 12p-A Clinical Study of 5 New Cases and a Literature Review". Genes. 12 (6): 811. doi:10.3390/genes12060811. PMC 8226674. PMID 34073526.
- 1 2 Polityko, A.D., E. Goncharova, L. Shamgina; et al. (2005). "Pallister-Killian Syndrome : Rapid Decrease of Isochromosome 12p Frequency during Amniocyte Subculturing. Conclusion for Strategy of Prenatal Cytogenetic Diagnostics". Journal of Histochemistry and Cytochemistry. 53 (3): 361–364. doi:10.1369/jhc.4A6402.2005. PMID 15750020.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Schubert R, Viersbach R, Eggermann T, Hansmann M, Schwanitz G (October 1997). "Report of two new cases of Pallister-Killian syndrome confirmed by FISH: tissue-specific mosaicism and loss of i(12p) by in vitro selection". American Journal of Medical Genetics. 72 (1): 106–10. doi:10.1002/(sici)1096-8628(19971003)72:1<106::aid-ajmg21>3.0.co;2-u. PMID 9295085.
- ↑ Wang T, Ren C, Chen D, Lu J, Guo L, Zheng L, Liu Y, Chen H (2019). "Prenatal diagnosis of Pallister-Killian syndrome using cord blood samples". Molecular Cytogenetics. 12: 39. doi:10.1186/s13039-019-0449-x. PMC 6717365. PMID 31497069.
- ↑ Salzano E, Raible SE, Kaur M, Wilkens A, Sperti G, Tilton RK, Bettini LR, Rocca A, Cocchi G, Selicorni A, Conlin LK, McEldrew D, Gupta R, Thakur S, Izumi K, Krantz ID (December 2018). "Prenatal profile of Pallister-Killian syndrome: Retrospective analysis of 114 pregnancies, literature review and approach to prenatal diagnosis". American Journal of Medical Genetics. Part A. 176 (12): 2575–2586. doi:10.1002/ajmg.a.40499. PMID 30289601. S2CID 52921433.
- ↑ "Trisomy 13: MedlinePlus Genetics". Archived from the original on 2021-07-09. Retrieved 2021-07-24.
- ↑ "Trisomy 18: MedlinePlus Genetics". Archived from the original on 2021-07-09. Retrieved 2021-07-24.
- ↑ "Prenatal cell-free DNA screening - Mayo Clinic". Archived from the original on 2021-07-23. Retrieved 2021-07-24.
- ↑ "Genome-Wide Association Studies Fact Sheet". Archived from the original on 2019-02-01. Retrieved 2021-07-24.
- ↑ Chau MH, Lam DY, Zhu X, Kwok YK, Ting YH, Chan WP, Shi M, Cheung SW, Lau TK, Ville Y, Leung TY, Choy KW (July 2020). "The utility of genome-wide cell-free DNA screening in the prenatal diagnosis of Pallister-Killian syndrome". Prenatal Diagnosis. 40 (8): 1005–1012. doi:10.1002/pd.5721. PMID 32350887. S2CID 217587371.
- ↑ Gasparini Y, Montenegro MM, Novo-Filho GM, Ceroni JR, Honjo RS, Zanardo ÉA, Dias AT, Nascimento AM, Costa TV, Madia FA, Chehimi SN, Damasceno JG, Kim CA, Kulikowski LD (2019). "Mosaic Trisomy 12 Associated with Overgrowth Detected in Fibroblast Cell Lines". Cytogenetic and Genome Research. 157 (3): 153–157. doi:10.1159/000498836. PMID 30933946. S2CID 91187498.
- ↑ Weiss K, Lachlan K, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A (1993). ""CHD4 Neurodevelopmental Disorder"". PMID 32881470.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Schubert, R., R. Viersbach, T. Eggermann (1997). "Report of two new cases of Pallister-Killian syndrome confirmed by FISH: tissue-specific mosaicism and loss of i(12p) by in vitro selection". Am J Med Genet. 72 (1): 106–110. doi:10.1002/(SICI)1096-8628(19971003)72:1<106::AID-AJMG21>3.0.CO;2-U. PMID 9295085.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Zambon, Francesco (2001-05-22). "Pallister-Killian syndrome". Medline Current Contents. Archived from the original on 2006-05-09. Retrieved 2006-05-31.
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Tetrasomy 12p". www.orpha.net. Archived from the original on 10 July 2021. Retrieved 9 August 2021.
External links
| Classification | |
|---|---|
| External resources |
|