داء ريفسام
داء رِيفسام أو مرض رِيفسام أو نقص إنزيم أكسيديز حمض الفيتانيك (بالإنجليزية: Refsum's disease) هو مرض عصبي وراثي متنحي، [1] ينتج عنه تراكم حمض الفيتانيك في الخلايا والأنسجة، ومعروف أيضاً باسم داء ريفسام الكلاسيكي أو الخاص بالبالغين.[2][3][4][5]
| داء ريفسام | |
|---|---|
داء ريفسام | |
| معلومات عامة | |
| الاختصاص | طب الجهاز العصبي |
| من أنواع | عسر شحميات الدم، ومرض |
وهو واحد من العديد من الاضطرابات التي سُميت بعد طبيب الأعصاب النرويجي سيغفالد برنارد ريفسوم (1907-1991).[6][7]
عادةً ما يتم تشخيص هذا المرض في بداية مرحلة المراهقة، عن طريق حمض الفيتانيك، حيث يكون فوق المستوى المتوسط. يحصل الأشخاص على حمض الفيتانيك اللازم في المقام الأول من خلال نظامهم الغذائي، على الرغم من أن وظيفة هذا الحمض الفيسيولوجية لا تزال مجهولة في البشر، لكنه يلعب دوراً في تنظيم التمثيل الغذائي للأحماض الدهنية في الكبد عند الفئران.[8]
الخصائص
الأفراد الذين يعانون من داء ريفسام والذي يتمثل في الضرر العصبي، انحطاط المخيخ، والاعتلال العصبي المحيطي. حيث يكون أكثر شيوعا في مرحلة الطفولة والمراهقة مع مسار تقدمي، على الرغم من فترات من الركود. وتشمل الأعراض أيضا ترنح، جلد متقشر (سماك)، وصعوبة في السمع، ومشاكل في العين بما في ذلك التهاب الشبكية الصباغي، إعتام عدسة العين، والعمى الليلي.[5] 80٪ من المرضى الذين تم تشخيصهم بداء ريفسام، تبين انهم يعانون فقدان السمع الحسي العصبي.[9] والذي يكون نتيجة للأضرار التي لحقت الأذن الداخلية أو العصب الذي يصل الأذن إلى الدماغ.
السبب
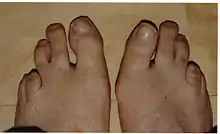
مرض ريفسام هو اضطراب البيروكسيسومات الناجم عن ضعف الأكسدة من نوع ألفا، لسلاسل الأحماض الدهنية المتفرعة، مما يؤدي إلى تراكم حمض الفيتانيك ومشتقاته في البلازما والأنسجة.
قد يكون هذا بسبب النقص في هيدروكسيلاز فنتانويل مرافق الإنزيم أ، أو نشاط بيروكسين 7.
بشكل عام، مرض ريفسام ناتج عن طفرات PHYH. [10][11]
يمكن للطفرات في الجينPEX7 أن تقطع النقل البيروكسيزومي للبروتينات، حيث أن هذا الجين يعطي مستقبل البروتين بيروكسين 7.
هذه الطفرات في الجين PEX7 تؤدي بشكل عام إلى خلل التنسج الغضروفي المنقط نوع 1، مما يؤدي إلى ضعف في النمو في أجزاء عديدة من الجسم. [12]
ويُرث مرض ريفسام، في نمط وراثي متنحي، أي أنه يتطلب حدوث الطفرة في كلا النسختين من الجين (كلا الأليلين) ليحدث.
التصنيف
يمكن تقسيم داء ريفسام عند البالغين إلى قسمين :
داء ريفسام الأول وداء ريفسام الثاني
الأول ينبع من الطفرات في هيدروكسيلاز فيتانويل- مرافق الإنزيم أ، على موقع 10p13 على كروموسوم [6q22-24. [11}[13] كان يعتقد في البداية أن هذه هي الطفرة الوحيدة؛ ولكن 55٪ من الحالات تعزى الآن إلى الطفرات في الجينات الأخرى. [5]
داء ريفسام 2 ينبع من الطفرات في جين البيروكسين 7 (PEX7). هذه الطفرة على الجين PEX7 هي أيضا على كروموسوم 6q22-24، ووجد في المرضى الذين يعانون من تراكم حمض الفيتانيك بدون طفرة PHYH.
لا ينبغي الخلط بداء ريفسام البالغين وداء ريفسام الطفلي، وهو اضطراب بيوكسيسوم بيوجينيسيس الناجم عن أوجه القصور في هدم الأحماض الدهنية طويلة السلسلة والأحماض الدهنية متفرعة السلسلة (مثل حمض الفيتانيك) والبلازمالوجين الحيوي[14][11]
العلاج
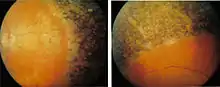
بما أن حمض الفيتانيك لا ينتج في جسم الإنسان، فإن الأفراد الذين يعانون من مرض ريفسام عادةً ما يوضعون في نظام غذائي خالٍ من حمض الفيتانيك، ويتجنبون استهلاك الدهون من الحيوانات المجترة، وبعض الأسماك كالتونا وأسماك القد. [15][16] كما عليهم أن يتجنبوا الحيوانات التي تتغذى على العشب وحليبها. [17]
أظهرت الأبحاث الحديثة أن إنزيمات CYP4 يمكن أن تساعد في تقليل تراكم حمض الفيتانيك في أجسام الكائنات الحية. [18]
يستخدم علاج المرضى طرق أخرى مثل البلازمافوريسيس (Plasmapheresis) أي استخراج البلازما - وهو تدخل طبي قائم على إزالة بلازما الدم من الجسم، عن طريق سحب الدم، وفصله إلى البلازما والخلايا، ونقل الخلايا مرة أخرى إلى مجرى الدم، وغالباً ما يستخدم لإزالة الأجسام المضادة في علاج أمراض المناعة الذاتية - حيث تستخدم هذه الطريقة لتصفية الدم لضمان عدم تراكم حمض الفيتانيك. [17]
المصادر البيولوجية لحمض الفيتانيك
في الحيوانات المجترة، تخمير الأمعاء للمواد النباتية المستهلكة يحرر الفيتول، وهو مكون من الكلوروفيل، والذي يتم تحويله بعد ذلك إلى حمض الفيتانيك وتخزينها في الدهون.[1] على الرغم من أن البشر لا يمكن أن تستمد كميات كبيرة من حمض الفيتانيك من استهلاك الكلوروفيل الموجودة في المواد النباتية، فقد اقترح أن القرود العليا (بونوبوس، الشمبانزي، الغوريلا، والأورانجوتان) يمكن أن تستمد كميات كبيرة من حمض الفيتانيك من تخميرالمواد النباتية.[2]
المراجع
- Verhoeven, N. M.; Wanders, R. J.; Poll-The, B. T.; Saudubray, J. M.; Jakobs, C. (1998). "The metabolism of phytanic acid and pristanic acid in man: a review". Journal of Inherited Metabolic Disease.
- Watkins, P. A.; Moser, A. B.; Toomer, C. B.; Steinberg, S. J.; Moser, H. W.; Karaman, M. W.; Ramaswamy, K.; Siegmund, K. D.; Lee, D. R.; Ely, J. J.; Ryder, O. A.; Hacia, J. G. (2010). "Identification of differences in human and great ape phytanic acid metabolism that could influence gene expression profiles and physiological functions". BMC Physiology.
- Fitzpatrick's dermatology in general medicine. (ط. 6th ed.)، New York: McGraw-Hill, Medical Pub. Division، 2003، ISBN 0071380760، OCLC 49526920، مؤرشف من الأصل في 18 ديسمبر 2019.
{{استشهاد بكتاب}}:|edition=has extra text (مساعدة) - 1950-, James, William D. (William Daniel), (2006)، Andrews' diseases of the skin : clinical dermatology. (ط. 10th ed.)، Philadelphia: Saunders Elsevier، ISBN 0721629210، OCLC 62736861، مؤرشف من الأصل في 18 ديسمبر 2019.
{{استشهاد بكتاب}}:|edition=has extra text (مساعدة)صيانة CS1: extra punctuation (link) صيانة CS1: أسماء عددية: قائمة المؤلفون (link) - Dermatology (ط. 2nd ed)، [St. Louis, Mo.]: Mosby/Elsevier، 2008، ISBN 9781416029991، OCLC 212399895، مؤرشف من الأصل في 18 ديسمبر 2019.
{{استشهاد بكتاب}}:|edition=has extra text (مساعدة) - Refsum S (1945)، "Heredoataxia hemeralopica polyneuritiformis - et tidligere ikke beskrevet familiært syndrom? En foreløbig meddelelse"، Nordisk Medicin (باللغة النرويجية)، 28: 2682–6.
- Refsum S (1946)، "Heredopathia atactica polyneuritiformis. A familial syndrome not hitherto described. A contribution to the clinical study of hereditary diseases of the nervous system"، Acta psych. neur. (Suppl.38): 1–303.
- Nogueira, C.؛ Meehan, T.؛ O’Donoghue, G. (2014)، "Refsum's Disease and Cochlear Implantation"، Annals of Otology, Rhinology, and Laryngology، 123 (6): 425–427، doi:10.1177/0003489414526846.
- Nogueira, C.; Meehan, T. & O’Donoghue, G. (2014). "Refsum's Disease and Cochlear Implantation". Annals of Otology, Rhinology, and Laryngology. 123 (6): 425–427. doi:10.1177/0003489414526846
- Online Mendelian Inheritance in Man (OMIM) 266500
- Online Mendelian Inheritance in Man (OMIM) 266510
- Lambert-Hamill, M.; Mitchell, J.; Wierzbicki, A. S.; Haasjes, J.; Brites, P.; Waterham, H. R.; et al. (2003). "Identification of PEX7 as the Second gene involved in Refsum disease". The American Journal of Human Genetics. 72 (2): 471–477. doi:10.1086/346093
- Lambert-Hamill, M.; Mitchell, J.; Wierzbicki, A. S.; Haasjes, J.; Brites, P.; Waterham, H. R.; et al. (2003). "Identification of PEX7 as the Second gene involved in Refsum disease". The American Journal of Human Genetics.
- Online Mendelian Inheritance in Man
- National Institutes of Health. "Synonym(s): Phytanic Acid Storage Disease, Heredopathia Atactica Polyneuritiformis <Internet>". Retrieved 8 July 2007.
- Baldwin, R. J.; et al. (2010). "The effectiveness of long-term dietary therapy in the treatment of adult Refsum disease". J Neurol Neurosurg Psychiatry. 81 (9): 954–957. doi:10.1136/jnnp.2008.161059. PMID 20547622.
- Metoyer, K. "Adult Refsum Disease. In C. Noggle". The encyclopedia of neuropsychological disorders.
- Xu F, Ng VY, Kroetz DL, de Montellano PR (2006). "CYP4 isoform specificity in the omega-hydroxylation of phytanic acid, a potential route to elimination of the causative agent of Refsum's disease". J. Pharmacol. Exp. Ther. 318 (2): 835–9. doi:10.1124/jpet.106.104976. PMID 16707724.
 بوابة طب
بوابة طب