متلازمة القلب الأيسر ناقص التنسج
متلازمة القلب الأيسر ناقص التنسج هو عيب خلقي نادر في القلب يكون فيه الجانب الأيسر من القلب غير متطور. قد يصيب البطين الأيسر، أو الأبهر، أو الصمام الأبهري، أو الصمام التاجي.[1]
| متلازمة القلب الأيسر ناقص التنسج | |
|---|---|
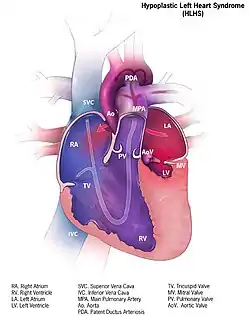 توضيح لمتلازمة القلب الأيسر ناقص التنسج توضيح لمتلازمة القلب الأيسر ناقص التنسج | |
| معلومات عامة | |
| الاختصاص | طب القلب |
| من أنواع | داء القلب الزراقي، ومرض قلبي خلقي، واعتلال قلبي وحيد البطين ، ومرض |
| الإدارة | |
| أدوية | |
العلامات والأعراض
تكون القناة الشريانية عند الولادة ما تزال مفتوحة، وتوجد لتدفق الدم إلى الرئتين مقاومة أعلى من الطبيعية. يسمح هذا بتزويد مقبول للأكسجين من خلال المزج بين الأذينين ويعطي المظهر الطبيعي عند الولادة. عندما تبدأ القناة بالانغلاق وتنقص مقاومة الأوعية الرئوية، يتوقف تدفق الدم عبر القناة ويزداد التدفق إلى الرئتين، مما ينقص وصول الأكسجين إلى الدورة الدموية الكبرى. قد يتسبب بحدوث زرقة وضائقة تنفسية يمكن أن تتطور إلى صدمة قلبية.[2]
يكون العرض الأول إما الزرقة التي لا تستجيب بإعطاء الأكسجين أو التغذية السيئة. قد يكون النبض المحيطي ضعيفًا والأطراف باردة عند لمسها.
يشيع حدوث إتش إل إتش إس عادة عند انخفاض وزن الولادة والولادة الباكرة.
يوجد عند الولدان المصابين بعيب حاجز أذيني صغير، يسمى «مقيد»، امتزاجًا غير مناسب بين الدم المؤكسج وغير المؤكسج. سرعان ما تتدهور حالة هؤلاء الولدان ويطورون حماض وزرقة.
على تخطيط كهربائية القلب، يشيع انحراف المحور الأيمن وتضخم البطين الأيمن، ولكنه ليس نوعي للإتش إل إتش إس. قد تظهر صورة الصدر الشعاعية البسيطة قلب كبير (تصخم قلب) أو زيادة في توعية الرئتين. لا يكون لدى الولادان المصابين بإتش إل إتش إس عادةً لغط القلب (نفخة قلبية). ولكن في بعض الحالات، قد تسمع نفخة جريان الصمام الرئوي أو نفخة قلس الصمام ثلاثي الشرف.
قد يتسبب الترافق مع قلس ثلاثي الشرف أو خلل وظيفة البطين الأيمن بتطور ضخامة كبدية.
الأسباب
لا يوجد سبب معروف في غالبية حالات إتش إل إتش إس.[3] فقد يوجد في بعض الحالات مكون وراثيًا، إذ أظهر إتش إل إتش إس قابلية للتوريث وارتباطه مع طفرات جينية محددة.[4][5]
الأسباب البيئية
في تحليل تراجعي لأكثر من 1300 مولود (مولودين بين عامي 1996 و2006) من 25 مشفى أطفال في الولايات المتحدة، وجد باحثون في مشفى سينسيناتي للأطفال في أوهايو أن الأطفال المصابين بالإتش إل إتش إس كانوا أكثر ولادة في أشهر الصيف، مما يشير إلى لعب العوامل الموسمية والبيئية دورًا مهمًا في حدوث الحالة.[6]
علم الوراثة
تتضمن المواقع الجينية المرتبطة بإتش إل إتش إس، GJA1 (كونيكسي 43)، وHAND1، وNKX2.5، و10q22، و6q23. هناك خطر خفيف لتكرار الحالة في الحمول القادمة، يقدر بنسبة 2-4% وتزداد في العائلات التي لديها حالتين مصابتين بنسبة 25%. يُعتقد وجود توسط لطفرات جينية ونفوذية (انتفاذ) غير مكتمل.[2][7][8]
يرتبط إتش إل إتش إس أيضًا مع العديد من المتلازمات الوراثية تشمل تثلث الصبغي 13 (متلازمة باتو)، تثلث الصبغي 18 (متلازمة إدوارد)، وتثلث الصبغي 9 جزئيًا، ومتلازمة تيرنر (إكس أو)، ومتلازمة جاكوبسن (متلازة حذف 11q)، متلازمة هولت-أورام، ومتلازمة سيمث-ليملي-أوبيتز.
عوامل الخطر
يزيد وجود ورم رطب كيسي من خطر الإصابة بإتش إل إتش إس عند الجنين.[9]
الفيزيولوجيا المرضية
في التشريح النموذجي، يتلقى الجانب الأيسر من القلب الدم الغني بالأكسجين من الرئتين ويضخه إلى باقي أنحاء الجسم. عند الأشخاص المصابين بإتش إل إتش إس، يكون الشريان الأبهري والبطين الأيسر غير متطورين (يبدأ من الرحم)، ويكون كلًّا من الصمامين الأبهري والتاجي صغيرين جدًا على السماح بجريان كافٍ أو رتقيين (منغلقين).[10] عندما يعود الدم من الرئتين إلى الأذين الأيسر، لا يمكن ضخه إلى باقي أنحاء الجسم من خلال البطين الأيسر.[11] يعتمد الوليد على جريان الدم من خلال عيب الحاجز الأذيني من أجل امتزاج الدم المؤكسج مع الدم غير المؤكسج، وعلى القناة الشريانية المفتوحة للسماح للدم بالوصول إلى الأبهر والدورة الدموية الكبرى من خلال البطين الأيمن. ولذلك يدعى إتش إل إتش إس بعيب «البطين المفرد».
التشخيص
يمكن تشخيص متلازمة القلب الأيسر ناقص التنسج قبل الولادة أو عند الولادة من خلال تخطيط صدى القلب. تتضمن الموجودات النموذجية بطين أيسر وأبهر صغيرين، وشذوذ الصمامين التاجي والأبهري، تدفق ارتجاعي في القوس المعترضة للأبهر، وتدفق أيسر-أيمن بين الأذينين. يُشاهد عادةً خلال الثلث الثاني من الحمل، بين الأسبوعين الحمليين 18 و24.[2]
التدبير
طبيًا
يكون إتش إل إتش إس مميتًا إذا لم تجرَ تدخلات تطيل الحياة، ولكن مع تدخلات فقد يعيش الوليد. يمكن إجراء سلسلة من عمليات جراحة القلب والصدر أو عملية زراعة قلب كامل. وبينما ظهر التدخل الجراحي على أنه وسيلة الرعاية المعيارية في الولايات المتحدة، تقارب نظم صحية وطنية، خاصة في فرنسا، إتش إل إتش إس بطرق أكثر تحفظًا، من خلال التأكيد على إنهاء الحمل أو رعاية رحيمة بعد الولادة.[12]
قبل الجراحة، يجب إبقاء القناة الشريانية مفتوحة للسماح بجريان الدم من خلال استخدام أدوية تتضمن البروستاغلاندينات. يُستخدم هواء أكسجينه أقل من الطبيعي عند الولدان المصابين بمتلازمة تضخم البطين الأيسر. تزيد مستويات الأكسجين المنخفضة من المقاومة الوعائية الرئوية (بّي في أر) فيتحسن جريان الدم إلى باقي الجسم بسبب الاختلاف الكبير في الضغط بين الرئتين والجسم. يتطلب الوصول إلى أكسجين أقل من الجو المحيط استخدام النتروجين المستنشق. يعد أحادي أكسيد النتروجين موسعًا وعائيًا رئويًا فعالًا، فيخفض بّي في أر ويحسن العود الوريدي. يمكن لأي عامل رافع للبّي في أر أن يعرقل جريان الجانب الأيمن.[13][14]
جراحيًا
تكون العمليات الجراحية المساعدة عند وجود تضخم قلب أيسر معقدة وتحتاج لأن تكون مخصصة حسب كل مريض. يجب أن يقيم طبيب القلب جميع الخيارات الطبية والجراحية على أساس كل حالة بحالتها.
في الوقت الحالي، يخضع الولدان لجراحة استنباتية (ترميمية) منظمة (إجراء سانو أو نوروود في الأيام الأولى بعد الولادة، إجراء غلين أو هيمي-فونتان بين الشهرين الثالث والسادس، وعملية فونتان بين السنة والنصف وحتى الخمس سنوات) أو زراعة قلب. تشير التوقعات الحالية إلى أن 70% من المرضى المصابين بإتش إل أتش إس يمكن أن يصلوا لعمر البلوغ.[15] تظهر العديد من الدراسات أنه كلما ارتفع حجم (عدد الإجراءات الجراحية) في المشفى، كلما كان معدل المراضة (الموت) أقل.[16][17] تتضمن العوامل التي تزيد الخطر على الولدان الوزن الناقص عند الولادة، أو الشذوذات الخلقية، أو المتلازمة الوراثية، أو المصابين بحاجز الأذين المقيد للغاية. تصل البقيا لخمس سنوات إلى ما يقارب 80% عند المرضى بدون عوامل خطر.[18] تبدي الدراسات أن نحو 75% من هؤلاء الأطفال الذين يعيشون بفضل الجراحة لديهم تأخرات تطورية في مجال واحد أو أكثر، مثل الحركي، أو الإدراكي، أو إعاقات لغوية، ويعاني نحو ثلث الأطفال ذوي البطين المفرد وبدون متلازمة وراثية من إعاقات مهمة.[19] تركز الأبحاث الحالية على تتبع العلاقات بين الإصابات التطورية العصبية، والإجراءت الجراحية ووحدة العناية المكثفة، والقابلية الوراثية بهدف تعديل التدخلات التي تتسبب بإعاقات تطورية عصبية ونفسية. تعد عملية هايبرد بديلًا عن عملية نوروود.[20]
يقدم بعض الأطباء «رعاية رحيمة»، بدلًا من الجراحات، التي تتسبب بموت الأطفال، عادة خلال أول أسبوعين بعد الولادة. يشرف الطبيب على الرعاية الرحيمة، ويمكن إجراؤها سواء في المشفى أو المنزل. على أي حال، نظرًا إلى التحسن الشاسع في التدخلات الجراحية، إذ حققت العديد من المستشفيات نسبة بقيا تصل إلى أكثر من 90%، فيدور نقاش حول فيما إذا يجب الاستمرار بعرض «الرعاية الرحيمة» على العائلات.[21] توصلت دراسة في عام 2003 أجريت على مجموعة من الأطباء الخبيرين بمجال رعاية الأطفال المصابين بإتش إل إتش إس إلى انقسام الأطباء بالتساوي عندما سُئلوا عما سيفعلونه إذا كان ابنهم مصاب بإتش إل إتش إس، إذ صرح 1/3 منهم باختيارهم الجراحة، و1/3 منهم باختيارهم للعلاج التلطيفي (الرحيم) بدون جراحة، و1/3 غير متأكدين حول ما سيقومون به.[22]
يعتبر الإجراء ثلاثي المراحل إجراءً تلطيفيًا (ليس شافيًا)، لأنه يصنع للأطفال دورة دموية تعمل في حجرتين فقط من حجرات القلب الأربع.
إجراء نوروود
إجراء نوروود هو الخطوة الأولى. في هذا الإجراء، يستخدم البطين الأيمن لضخ الدم إلى الدورة الدموية الكبرى. ونظرًا لأن القلب الأيمن لا يضخ الدم مباشرة إلى الرئتين، تجرى تحويلة من أجل عبور الدم غير المؤكسج خلال الرئتين. إما من خلال وصل الشريان تحت الترقوة مع الدورة الدموية الصغرى (تحويلة بلالوك-توسيغ)، أو تحويلة مباشرة من البطين الأيمن إلى الدورة الدموية الصغرى (تحويلة سانو). يُوسع الأبهر المتضيق باستخدام رقعة لتحسين الجريان الدموي في الجسم.[23]
قد يصبح الطفل خلال هذه الفترة ضعيفًا طبيًا وتحصل لديه مشاكل في التغذية لأن القلب يعمل بصعوبة للغاية. توجد درجة هامة من الامتزاج الوريدي في البطين الأيمن، مما يؤدي إلى إشباع أكسجيني أقل. بالإضافة إلى هذا، تعرض التحويلتان بلالوك-توسيغ وسانو الرئتين للضغط الشرياني الجهازي، مما يتسبب بارتفاع ضغط رئوي طويل الأمد وقصور قلب في نهاية المطاف.[24]
إجراء هايبرد
قد يُستخدم إجراء هايبرد عوضًا عن نوروود.[24][25][26] لا يتطلب إجراء هايبرد استخدام مجازة قلبية رئوية أو إجراء بضع قص. عوضًا عن الجراحة لمدة ست ساعات، يأخذ إجراء هايبرد عادةً ساعة إلى ساعتين. في هذا الإجراء، توضع دعامة في القناة الشريانية للمحافظة على سالكيتها، وتوضع أشرطة على فرعي الشريان الرئوي الأيمن والأيسر للحد من الضغط والدورة الدموية المفرطة على الرئتين.[27] تكون نتائح طريقة هايبرد متطابقةً مع نتائج نوروود.
إجراء غلين
تخفف المرحة الثانية -إجراء غلين ثنائي الاتجاه أو هيمي-فونتان (انظر أيضًا إلى إجراء كاواشيما)- بعض المشاكل الحاصلة من تلطيف المرحلة الأولى. في هذه العملية، يُربط وريد الأجوف العلوي من ناحية القلب ويُوصل بالدورة الدموية الصغرى. خلال هذه المرحلة، تتفكك تحويلة بلالوك-توسيغ أو تحويلة سانو. لم تعد الرئتان تتعرضان للضغط الشرياني الجهازي، بل لضغط وريدي أقل. وكذلك لم يعد الدم الوريدي القادم من النصف العلوي للجسم يختلط مع الدم المؤكسج في البطين الأيمن، يبقى هنالك اختلاط وريدي من النصف السفلي للجسم، مما يؤدي إلى درجة من إزالة الإشباع الأكسجيني.[24]
إجراء فونتان
يكمل الإجراء الأخير، إجراء فونتان، إصلاح القلب الأيمن ناقص التصنع. على الرغم من وجود اختلافات عديدة، يكون التأثير الوظيفي بإعادة توجيه الدم الوريدي القادم من النصف السفلي من الجسم (عبر وريد الأجوف السفلي) بعيدًا عن الأذين الأيمن نحو الشريان الرئوي. يجب أن ينهي هذا أي امتزاج للدم المؤكسج مع الدم غير المؤكسج في البطين الأيمن. يقوم البطين الأيمن بالوظيفة التقليدية للبطين الأيسر، فيزوّد الجسم بالدم المؤكسج، بينما يقوم الضغط الوريدي الجهازي السلبي بوظيفة البطين الأيمن، عن طريق توصيل الدم غير المؤكسج إلى الرئتين.
الجراحة الجنينية
يُستقصى حاليًا عن إجراء التداخلات خلال التطور الجنيني. يُجرب إجراء فغر للحاجز بين الأذينين بطريق الجلد، عند الأجنة الذين لديهم تضخم في البطين الأيسر وحاحز بين الأذينين سليم.
المراجع
- Tchervenkov, C. I؛ Jacobs, J. P؛ Weinberg, P. M؛ Aiello, V. D؛ Béland, M. J؛ Colan, S. D؛ Elliott, M. J؛ Franklin, R. C؛ Gaynor, J. W؛ Krogmann, O. N؛ Kurosawa, H؛ Maruszewski, B؛ Stellin, G (2006)، "The nomenclature, definition and classification of hypoplastic left heart syndrome"، Cardiology in the Young، 16 (4): 339–368، doi:10.1017/s1047951106000291، PMID 16839428.
- Fulton, David R. (26 أكتوبر 2017)، "Hypoplastic left heart syndrome"، Up To Date، مؤرشف من الأصل في 23 ديسمبر 2019، اطلع عليه بتاريخ 30 نوفمبر 2017.
- Barron, D. J., Kilby, M. D., Davies, B., Wright, J. G., Jones, T. J., & Brawn, W. J. (2009)، "Hypoplastic left heart syndrome"، The Lancet، 374 (9689): 551–564، doi:10.1016/s0140-6736(09)60563-8، PMID 19683641.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Hinton, R. B., Martin, L. J., Tabangin, M. E., Mazwi, M. L., Cripe, L. H., & Benson, D. W. (2007)، "Hypoplastic left heart syndrome is heritable"، Journal of the American College of Cardiology، 50 (16): 1590–1595، doi:10.1016/j.jacc.2007.07.021، PMID 17936159.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Dasgupta C, Martinez AM, Zuppan CW, Shah MM, Bailey LL, Fletcher WH (2001)، "Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE)"، Mutat. Res.، 479 (1–2): 173–86، doi:10.1016/S0027-5107(01)00160-9، PMID 11470490.
- Eghtesady P, Brar A, Hall M (فبراير 2011)، "Seasonality of hypoplastic left heart syndrome in the United States: a 10-year time-series analysis"، J. Thorac. Cardiovasc. Surg.، 141 (2): 432–8، doi:10.1016/j.jtcvs.2010.06.060، PMID 20817208.
- Feinstein, JA؛ Benson, DW؛ Dubin, AM؛ Cohen, MS؛ Maxey, DM؛ Mahle, WT؛ Pahl, E؛ Villafañe, J؛ Bhatt, AB؛ Peng, LF؛ Johnson, BA؛ Marsden, AL؛ Daniels, CJ؛ Rudd, NA؛ Caldarone, CA؛ Mussatto, KA؛ Morales, DL؛ Ivy, DD؛ Gaynor, JW؛ Tweddell, JS؛ Deal, BJ؛ Furck, AK؛ Rosenthal, GL؛ Ohye, RG؛ Ghanayem, NS؛ Cheatham, JP؛ Tworetzky, W؛ Martin, GR (03 يناير 2012)، "Hypoplastic left heart syndrome: current considerations and expectations"، Journal of the American College of Cardiology، 59 (1 Suppl): S1–42، doi:10.1016/j.jacc.2011.09.022، PMC 6110391، PMID 22192720.
- Hinton, R. B., Martin, L. J., Rame-Gowda, S., Tabangin, M. E., Cripe, L. H., & Benson, D. W. (2009)، "Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve"، Journal of the American College of Cardiology، 53 (12): 1065–1071، doi:10.1016/j.jacc.2008.12.023، PMC 2703749، PMID 19298921.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Cunningham, F. Gary؛ Leveno, Kenneth J.؛ Bloom, Steven L.؛ Spong, Catherine Y.؛ Dashe, Jodi S.؛ Hoffman, Barbara L.؛ Casey, Brian M.؛ Sheffield, Jeanne S. (2013)، "Fetal Imaging"، Williams Obstetrics (ط. 24)، New York, NY: McGraw-Hill Education.
- Galindo, A., Nieto, O., Villagrá, S., Grañeras, A., Herraiz, I., & Mendoza, A. (2009)، "Hypoplastic left heart syndrome diagnosed in fetal life: associated findings, pregnancy outcome and results of palliative surgery"، Ultrasound in Obstetrics & Gynecology، 33 (5): 560–566، doi:10.1002/uog.6355، PMID 19367583.
{{استشهاد بدورية محكمة}}: صيانة CS1: أسماء متعددة: قائمة المؤلفون (link) - Burns, Paul Burns and Jasper، "Hypoplastic Left Heart Syndrome | Congenital Heart Disease - Cove Point Foundation | Johns Hopkins Children's Hospital"، www.pted.org، مؤرشف من الأصل في 02 أبريل 2017، اطلع عليه بتاريخ 30 نوفمبر 2017.
- Noseda C, Mialet-Marty T, Basquin A (أبريل 2012)، "Hypoplasies sévères du ventricule gauche : soins palliatifs après un diagnostic prénatal"، Archives de pediatrie، 19 (4): 374–380، doi:10.1016/j.arcped.2012.01.022، PMID 22397767.
- Khambadkone S.؛ Li J.؛ De Leval M. R.؛ Cullen S.؛ Deanfield J. E.؛ Redington A. N. (2003)، "Basal pulmonary vascular resistance and nitric oxide responsiveness late after Fontan-type operation"، Circulation، 107 (25): 3204–3208، doi:10.1161/01.cir.0000074210.49434.40، PMID 12821557.
- Norwood W. I. (1991)، "Hypoplastic left heart syndrome. The"، Annals of Thoracic Surgery، 52 (3): 688–695، doi:10.1016/0003-4975(91)90978-y، PMID 1898174.
- Hypoplastic Left Heart Syndrome (HLHS) | The Children's Hospital of Philadelphia نسخة محفوظة 14 أكتوبر 2014 على موقع واي باك مشين.
- McHugh, KE؛ Hillman, DG؛ Gurka, MJ؛ Gutgesell, HP (يناير–فبراير 2010)، "Three-stage palliation of hypoplastic left heart syndrome in the University HealthSystem Consortium"، Congenital Heart Disease، 5 (1): 8–15، doi:10.1111/j.1747-0803.2009.00367.x، PMID 20136852.
- Hirsch, JC؛ Gurney, JG؛ Donohue, JE؛ Gebremariam, A؛ Bove, EL؛ Ohye, RG (يوليو 2008)، "Hospital mortality for Norwood and arterial switch operations as a function of institutional volume"، Pediatric Cardiology، 29 (4): 713–7، doi:10.1007/s00246-007-9171-2، PMID 18080151.
- Vojtovič, P.؛ Tláskal, T.؛ Gebauer, R.؛ Reich, O.؛ Chaloupecký, V.؛ Tomek, V.؛ Krupičková, S.؛ Matějka, T.؛ Hecht, P. (ديسمبر 2014)، "Long-term results of children operated for hypoplastic left heart syndrome in Children's Heart Centre"، Cor et Vasa، 56 (6): e449–e455، doi:10.1016/j.crvasa.2014.07.006، ISSN 0010-8650.
- Wernovsky, Gil؛ Licht, Daniel J. (2016)، "Neurodevelopmental Outcomes in Children with Congenital Heart Disease – What can we impact?"، Pediatric Critical Care Medicine، 17 (8 Suppl 1): S232–S242، doi:10.1097/PCC.0000000000000800، ISSN 1529-7535، PMC 4975480، PMID 27490605.
- Yabrodi, Mouhammad؛ Mastropietro, Christopher W. (04 أكتوبر 2016)، "Hypoplastic left heart syndrome: from comfort care to long-term survival"، Pediatric Research (باللغة الإنجليزية)، 81 (1–2): 142–149، doi:10.1038/pr.2016.194، ISSN 0031-3998، PMC 5313512، PMID 27701379.
- Wernovsky, Gil (01 سبتمبر 2008)، "The Paradigm Shift Toward Surgical Intervention for Neonates With Hypoplastic Left Heart Syndrome"، Archives of Pediatrics & Adolescent Medicine، 162 (9): 849–54، doi:10.1001/archpedi.162.9.849، PMID 18762602.
- Kon, Alexander A.؛ Ackerson, Lynn؛ Lo, Bernard (31 مايو 2003)، "Choices physicians would make if they were the parents of a child with hypoplastic left heart syndrome"، The American Journal of Cardiology، 91 (12): 1506–1509، doi:10.1016/S0002-9149(03)00412-0، PMID 12804748.
- "new norwood.gif"، مؤرشف من الأصل في 24 نوفمبر 2010.
- Feinstein, Jeffrey A.؛ Benson, D. Woodrow؛ Dubin, Anne M.؛ Cohen, Meryl S.؛ Maxey, Dawn M.؛ Mahle, William T.؛ Pahl, Elfriede؛ Villafañe, Juan؛ Bhatt, Ami B. (يناير 2012)، "Hypoplastic Left Heart Syndrome"، Journal of the American College of Cardiology، 59 (1): S1–S42، doi:10.1016/j.jacc.2011.09.022، PMC 6110391، PMID 22192720.
- Murphy, Michael O.؛ Bellsham-Revell, Hannah؛ Morgan, Gareth J.؛ Krasemann, Thomas؛ Rosenthal, Eric؛ Qureshi, Shakeel A.؛ Salih, Caner؛ Austin, Conal B.؛ Anderson, David R. (2015)، "Hybrid Procedure for Neonates With Hypoplastic Left Heart Syndrome at High-Risk for Norwood: Midterm Outcomes"، The Annals of Thoracic Surgery، 100 (6): 2286–2292، doi:10.1016/j.athoracsur.2015.06.098، PMID 26433522.
- Chauhan, Monika؛ Mastropietro, Christopher W. (2014)، "Hypoplastic Left Heart Syndrome in the Emergency Department: An Update"، The Journal of Emergency Medicine، 46 (2): e51–e54، doi:10.1016/j.jemermed.2013.08.061، PMID 24188609.
- Children's Hospital Boston | Pediatric Views نسخة محفوظة 29 فبراير 2012 على موقع واي باك مشين.
{kind=link}
{kind=link}
 بوابة طب
بوابة طب