متلازمة تريتشر كولينز
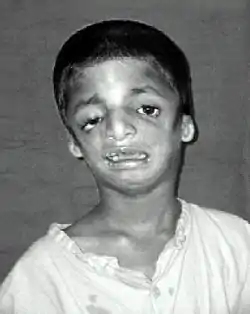
متلازمة تريتشر كولينز هي اضطراب وراثي يتميز بتشوهات في الأذنين، والعينين، وعظام الوجنتين، والذقن. تختلف شدّة المرض عند المصابين من خفيفة إلى شديدة. قد تشمل المضاعفات مشاكل في التنفس، والرؤية، والحنك المشقوق، وفقدان السمع. يمتلك المصابون بالمتلازمة عمومًا مستوى ذكاء متوسط.[1]
| متلازمة تريتشر كولينز | |
|---|---|
 متلازمة تريتشر كولينز | |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | مرض وراثي سائد ، ومتلازمة القوس الأول، ومتلازمة، ومرض |
وراثة المتلازمة من النوع الصبغي الجسدي المسيطر. تحدث في أكثر من نصف الحالات نتيجة طفرة جديدة بدلًا من أن تكون موروثة من والدي المصاب. قد تشمل الجينات المتورّطة TCOF1) أو POLR1C أو POLR1D)، يُشتبه بالتشخيص عمومًا بناءً على الأعراض ونتائج الأشعة السينية، وتُؤكَّد الإصابة عن طريق الفحص الجيني. [2]
متلازمة تريتشر كولينز غير قابلة للشفاء، لكن يمكن التحكم بالأعراض عن طريق الجراحة الترميمية، وأجهزة المساعدة السمعية، وعلاج النطق، والأجهزة المساعدة الأخرى.[3] متوسط العمر المتوقع طبيعي بشكل عام. تحدث متلازمة تريتشر كولينز عند حوالي واحد من بين 50,000 شخص. سميت المتلازمة تيمّنًا بالجراح وطبيب العيون الإنكليزي إدوارد تريشر كولينز، الذي وصف سمات المتلازمة الأساسية عام 1900.[4][5]
الأعراض والعلامات
تختلف الأعراض لدى الأشخاص المصابين بمتلازمة تريتشر كولينز، إذ يتأثر بعض الأفراد بشكل خفيف وتبقى المتلازمة غير مُشخَّصة لديهم، بينما يعاني البعض الآخر من أعراض معتدلة إلى شديدة مثل تشوّهات في الوجه تهدّد مجرى الهواء، ما يعتبر مهدّدًا للحياة لديهم.[6] معظم مظاهر متلازمة تريتشر كولينز متناظرة ويمكن التعرّف عليها عند الولادة.
أكثر أعراض متلازمة تريتشر كولينز شيوعًا هو التطوّر غير الكامل للفك السفلي وعظم الوجنة. يمكن أن يترافق ما سبق بانكماش اللسان. يمكن أن يؤدي صغر الفك السفلي إلى سوء إطباق الأسنان أو في الحالات الأكثر خطورة، صعوبة في التنفس أو البلع. يتركّز الاهتمام الأكبر على الجهاز التنفسي للطفل المصاب بمتلازمة تريتشر كولينز، وتُعالج المشاكل الأخرى بعد معالجة مشاكل الجهاز التنفسي.[7] يعطي التطوّر غير الكامل للعظم الوجني الخدين مظهرًا غائرًا. [8][9]
تكون الأذن الخارجية في بعض الأحيان صغيرة، أو مستديرة، أو مشوهة، أو غائبة تمامًا لدى الأشخاص المصابين بـمتلازمة تريتشر كولينز. وُصف تضيّق ثنائي الجانب ومتناظر في مجرى السمع الخارجي أو غياب تامّ في الأخير.[10] في معظم الحالات، تكون عظام الأذن الوسطى وتجويف الأذن الوسطى مشوهة. نادرًا ما وُصفَت تشوهات الأذن الداخلية. نتيجة لهذه التشوهات، يعاني أغلبية الأفراد المصابين بـمتلازمة تريتشر كولينز من فقدان السمع التوصيلي. [11][9]
يعاني معظم الأشخاص المصابين أيضًا من مشاكل في العين، بما في ذلك ثلامة (ثلم) الجفن السفلي، والغياب الجزئي أو الكامل للرموش على الجفن السفلي، والجفون ذات الزوايا السفلية، وإطراق الجفون العلوية والسفلية، وتضيق القناة الدمعية. يمكن أن يحدث فقدان الرؤية ويترافق مع الحول، وأخطاء وتفاوت الانكسار، ويمكن أن يحدث أيضًا بسبب جفاف العين الشديد، نتيجة لتشوهات الجفن السفلي والتهابات العين المتكررة.[12][13]
على الرغم من أن الجمجمة ذات الشكل غير الطبيعي ليست مميزة لمتلازمة تريتشر كولينز، ولكن لوحظ بعض الأحيان وجود قصر في الرأس وتضيّق في المسافة بين الصدغين. الحنك المشقوق شائع أيضًا.
تشاهد حالات شذوذ الأسنان عند 60% من الأشخاص المصابين، بما في ذلك عدم تكوّن الأسنان (33%)، وتغير لون السن (عتامة المينا) (20%)، وسوء توضّع الرحى الفكّية الأولى (13%)، والتباعد الواسع بين الأسنان. في بعض الحالات، تؤدي حالات شذوذ الأسنان مع نقص تنسج الفك السفلي إلى سوء الإطباق، ما يؤدي إلى مشاكل في تناول الطعام والقدرة على إغلاق الفم.
من الميزات الأقل شيوعًا لمتلازمة تريتشر كولينز التي تُضاف إلى المشاكل التنفسية لدى الشخص المصاب -بما في ذلك توقف التنفس أثناء النوم-رتق قمع الأنف (تضيق أو عدم وجود المنعرَين). تطوّر البلعوم غير الكامل يمكن أن يضيق مجرى الهواء أيضًا.
تشمل الميزات المرتبطة بمتلازمة تريتشر كولينز التي تُرى بشكل أقل تكرارًا: التشوهات الأنفية، والحنك عالي القوس، وتضخّم الشدق، وسوء توضّع الأشعار أمام صيوان الأذن، والحنك المشقوق، وفرط التباعد، والجفن العلوي المثلوم، وعيوب القلب الخلقية.
على الرغم من أن تشوه الوجه يرتبط غالبًا بتأخر النمو والإعاقة الذهنية، يمتلك أكثر من 95% من الأشخاص المصابين بمتلازمة تريتشر كولينز مستوى ذكاء طبيعي. يمكن أن تؤثر المشاكل النفسية والاجتماعية المرتبطة بتشوه الوجه على جودة الحياة لدى الأفراد المصابين بـالمتلازمة.
المعالجة
قد يشمل علاج الأفراد المصابين بـمتلازمة تريتشر كولنز تدخّل اختصاصات متعددة. يتركّز الاهتمام الأكبر على التنفس والتغذية، نتيجة نقص تنسج الفك السفلي وانسداد البلعوم السفلي باللسان. قد يحتاج الأطباء إلى ثقب القصبة الهوائية للحفاظ على مجرى هوائي مناسب، وفغر المعدة لضمان تناول السعرات الحرارية الكافية مع حماية مجرى الهواء.[14] تُجرى الجراحة التصحيحية للوجه في أعمار محددة، اعتمادًا على حالة النمو.[15]
لمحة عامة عن المبادئ التوجيهية الحالية:
- إذا كان الحنك المشقوق موجودًا، تُجرى الجراحة عادة في عمر 9-12 شهرًا. قبل الجراحة يجب إجراء تخطيط نوم مع وجود الصفيحة الحنكية، ما قد يتنبأ بحالة ما بعد الجراحة وإمكانية وجود توقف التنفس أثناء النوم بعد العملية. [16][17]
- يُعالَج فقدان السمع عن طريق تضخيم التوصيل عبر العظام، وعلاج النطق، والتعليم لتجنب مشاكل اللغة والكلام. تعد المعينات السمعية المثبتة على العظام بديلًا للأفراد الذين يعانون من تشوهات الأذن. [18]
- تُجرى عمليات إعادة البناء الوجني والحجاجي عندما يكتمل تطوّر العظام القحفية الحجاجية، وعادة في سن 5-7سنوات. تستخدم الطعوم العظمية الذاتية غالبًا عند الأطفال. يمكن استخدام المواد المالئة الشحمية في المنطقة المحيطة بالعضلة للحصول على نتيجة مثالية، وتُصحَّح ثلامة الجفن السفلي عن طريق سديلة جلدية عضلية تُرفَع لتغلق العيب. [19]
- عادة ما يُعاد بناء الأذن الخارجية عندما يكون عمر الفرد ثماني سنوات على الأقل. في بعض الأحيان، يمكن أيضًا علاج مشاكل القناة السمعية الظاهرة أو الأذن الوسطى.
- العمر الأمثل لإعادة بناء الفك السفلي مثير للجدل. اعتبارًا من عام 2004، استخدم هذا التصنيف:
- النوع الأول (معتدل) والنوع الثاني (متوسط) من 13 إلى 16 عامًا
- النوع الثاني-ب (تشوه متوسط إلى شديد) عند النضج الهيكلي
- النوع الثالث (شديد) 6-10 سنوات
يجرى التداخل على الأسنان تحت إشراف أخصائي تقويم الأسنان للتأكد من عدم حدوث تشوهات. إذا شوهدت تشوهات مثل سوء الإطباق أو فرط نمو الأسنان، يمكن اتخاذ الإجراءات المناسبة في أقرب وقت ممكن.
عادةً ما تطبَّق العلاجات المقوِّمة للفكين بعد سن 16 عامًا، إذ تكون كل الأسنان في مكانها والفك ومجموعة الأسنان الطبيعية ناضجة. عندما يُكتشَف وجود انقطاع النفس الانسدادي النومي، يُحدَّد مستوى الانسداد من خلال التنظير الداخلي للمسالك الهوائية العليا. يمكن أن يكون تقديم الفك السفلي طريقة فعالة لتحسين كل من التنفس والناحية الجمالية، في حين أن جراحة الذقن تستعيد الشكل فقط.[19]
إذا كان ترميم الأنف ضروريًا، يُجرى عادةً بعد الجراحة المقوِّمة للفكين وبعد سن 18 عامًا.
المراجع
- "Treacher Collins syndrome"، Genetics Home Reference (باللغة الإنجليزية)، يونيو 2012، مؤرشف من الأصل في 26 يونيو 2020، اطلع عليه بتاريخ 07 نوفمبر 2017.
- "Treacher Collins Syndrome"، NORD (National Organization for Rare Disorders)، 2016، مؤرشف من الأصل في 20 ديسمبر 2019، اطلع عليه بتاريخ 07 نوفمبر 2017.
- "Treacher Collins syndrome"، rarediseases.info.nih.gov (باللغة الإنجليزية)، 2015، مؤرشف من الأصل في 16 أغسطس 2019، اطلع عليه بتاريخ 07 نوفمبر 2017.
- Beighton, Greta (2012)، The Man Behind the Syndrome، Springer Science & Business Media، ص. 173، ISBN 9781447114154، مؤرشف من الأصل في 12 أغسطس 2020.
- R, Pramod John؛ John, Pramod (2014)، Textbook of Oral Medicine، JP Medical Ltd، ص. 76، ISBN 9789350908501، مؤرشف من الأصل في 12 سبتمبر 2020.
- Edwards, S J؛ Fowlie, A؛ Cust, M P؛ Liu, D T؛ Young, I D؛ Dixon, M J (01 يوليو 1996)، "Prenatal diagnosis in Treacher Collins syndrome using combined linkage analysis and ultrasound imaging."، Journal of Medical Genetics، 33 (7): 603–606، doi:10.1136/jmg.33.7.603، PMC 1050672، PMID 8818950.
- Mouthon, L., Busa, T., Bretelle, F., Karmous‐Benailly, H., Missirian, C., Philip, N., & Sigaudy, S. (2019). Prenatal diagnosis of micrognathia in 41 fetuses: Retrospective analysis of outcome and genetic etiologies. American Journal of Medical Genetics Part A, 179(12), 2365–2373. doi: 10.1002/ajmg.a.61359
- Katsanis SH, et al., Treacher Collins syndrome, 2004, GeneReviews
- "The Physician's Guide to Treacher Collins Syndrome" (PDF)، National Organization for Rare Disorders (NORD)، 2012، مؤرشف من الأصل (PDF) في 17 مايو 2017.
- Posnick, Jeffrey C (01 أكتوبر 1997)، "Treacher Collins syndrome: Perspectives in evaluation and treatment"، Journal of Oral and Maxillofacial Surgery، 55 (10): 1120–1133، doi:10.1016/S0278-2391(97)90294-9، PMID 9331237.
- Trainor, Paul A؛ Dixon, Jill؛ Dixon, Michael J (24 ديسمبر 2008)، "Treacher Collins syndrome: etiology, pathogenesis and prevention"، European Journal of Human Genetics، 17 (3): 275–283، doi:10.1038/ejhg.2008.221، PMC 2986179، PMID 19107148.
- Hertle, R W؛ Ziylan, S؛ Katowitz, J A (01 أكتوبر 1993)، "Ophthalmic features and visual prognosis in the Treacher-Collins syndrome."، British Journal of Ophthalmology، 77 (10): 642–645، doi:10.1136/bjo.77.10.642، PMC 504607، PMID 8218033.
- Marszałek, B؛ Wójcicki, P؛ Kobus, K؛ Trzeciak, WH (2002)، "Clinical features, treatment and genetic background of Treacher Collins syndrome."، Journal of Applied Genetics، 43 (2): 223–33، PMID 12080178.
- Goel L؛ وآخرون (2009)، "Treacher Collins syndrome-a challenge for anaesthesiologists"، Indian J Anaesth، 53 (4): 642–645، PMC 2894488، PMID 20640217.
- Evans, Adele Karen؛ Rahbar, Reza؛ Rogers, Gary F.؛ Mulliken, John B.؛ Volk, Mark S. (31 مايو 2006)، "Robin sequence: A retrospective review of 115 patients"، International Journal of Pediatric Otorhinolaryngology، 70 (6): 973–980، doi:10.1016/j.ijporl.2005.10.016، PMID 16443284.
- Bannink, Natalja؛ Mathijssen, Irene M.J.؛ Joosten, Koen F.M. (01 سبتمبر 2010)، "Use of Ambulatory Polysomnography in Children With Syndromic Craniosynostosis"، Journal of Craniofacial Surgery، 21 (5): 1365–1368، doi:10.1097/SCS.0b013e3181ec69a5، PMID 20856022، S2CID 43739792.
- Rose, Edmund؛ Staats, Richard؛ Thissen, Ulrike؛ Otten, Jörg-Eland؛ Schmelzeisen, Rainer؛ Jonas, Irmtrud (01 أغسطس 2002)، "Sleep-Related Obstructive Disordered Breathing in Cleft Palate Patients after Palatoplasty"، Plastic and Reconstructive Surgery، 110 (2): 392–396، doi:10.1097/00006534-200208000-00002، PMID 12142649، S2CID 36499038.
- Marres, HA (2002)، Hearing loss in the Treacher-Collins syndrome.، Advances in Oto-rhino-laryngology، ج. 61، ص. 209–15، doi:10.1159/000066811، ISBN 978-3-8055-7449-5، PMID 12408086.
- Zhang, Zhiyong؛ Niu, Feng؛ Tang, Xiaojun؛ Yu, Bing؛ Liu, Jianfeng؛ Gui, Lai (01 سبتمبر 2009)، "Staged Reconstruction for Adult Complete Treacher Collins Syndrome"، Journal of Craniofacial Surgery، 20 (5): 1433–1438، doi:10.1097/SCS.0b013e3181af21f9، PMID 19816274، S2CID 44847925.
 بوابة طب
بوابة طب