مرض شاركو-ماري-توث
مرض شاركو-ماري-توث أو اعتلال شاركو-ماري-توث العصبي، اعتلال الحركة والحس العصبي الموروث أو الضمور العضلي الشظوي هو مجموعة من الاضطرابات الموروثة المتغايرة جينياً وسريرياً تصيب الجهاز العصبي المحيطي ويتمثل بفقد مُتَرَقِّ في النسيج العضلي وحس اللمس في مختلف أجزاء الجسم دون التأثير على النمو العقلي أو أن يشكل خطراً على حياة المصاب. تم اكتشافه من قبل ثلاثة أطباء سنة 1886 وسمّي بأسمائهم. هذا المرض هو أحد أمراض الاعتلالات العصبية الأكثر انتشاراً ويصيب 1 من 2500 حالة ولادة.[2][3] تتلخص العلة في إصابة الأعصاب المحيطية التي تربط النخاع الشوكي بالعضلات مما يؤثر على نقل التدفُّعات العصبية ويؤدي إلى صعوبة في المشي واعوجاج في الرجلين منذ الطفولة المبكرة مع إمكانية تأخر ظهور المرض إلى فترة المراهقة. كان يُعدّ سابقاً أحد أنواع الحَثل العضلي.لا يتوفر حالياً شفاء من هذا المرض.
| مرض شاركو-ماري-توث | |
|---|---|
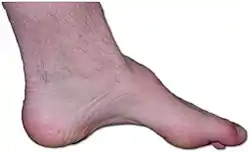 قدم شخص مصاب بمرض شاكو-ماري توث. يظهر ضعف العضلات،وتقوس الرجل،وإصبع قدم مخلبية هي علامات هذا المرض الوراثي. قدم شخص مصاب بمرض شاكو-ماري توث. يظهر ضعف العضلات،وتقوس الرجل،وإصبع قدم مخلبية هي علامات هذا المرض الوراثي. | |
| معلومات عامة | |
| الاختصاص | طب الجهاز العصبي |
| من أنواع | اعتلال عصبي حركي وحسي موروث، واضطراب عصبي عضلي، ومرض |
| الأسباب | |
| الأسباب | طفرة[1] |
| المظهر السريري | |
| الأعراض | وهن عضلي[1]، وضمور عضلي[1]، وقدم جوفاء[1]، وفقد الحس[1]، وضعف المنعكسات[1]، وانحراف العمود الفقري جانبيا[1] |
| الوبائيات | |
| انتشار المرض | 0.0003 [1] |
| التاريخ | |
| سُمي باسم | جان مارتن شاركو، وبيير ماري |
الأعراض والعلامات
عادةً ما يبدأ ظهور أعراض شاركو-ماري-توث مبكراً أثناء الطفولة أو عند البلوغ وقد تظهر أبكر من ذلك، لكن بعض الأشخاص لا يشعرون بالأعراض إلا في بداية الثلاثينيات أو الأربعينيات من العمر.تتمثل الأعراض الأوليّة غالباً بسقوط القدم (سقوط مقدم القدم بسبب ضعف، تهيّج أو ضرر للعصب الشظوي الأصليCommon peroneal nerve، أو شلل في عضلات الجزء الأمامي للساق، يفقد بسببها حركة الانثناء الظهراني للقدم). ويمكن أن تسبب أيضاً إصبع القدم المطرقية (انحناء الأصابع للأسفل).ضمور العضلات في الجزء السفلي من الساق قد يؤدي إلى مظهر "أقدام اللقلق". مع تقدم المرض، يعاني الكثير من المرضى من ضعف في عضلات اليد والساعد. يحدث فقدان لحس اللمس في القدمين والكاحلين والساقين، وكذلك في اليدين والرسغين والذراعين في أنواع مختلفة من المرض. ويكون في الأشكال المبكرة أو المتأخرة بسبب انقباضات عضلية تشنّجية على نحو متقطع والتي تجعل المريض عاجزاً عندما يُنشَّط المرض. القدم الجوفاء (قدم عالية التقوّس) أو القدم الرَحَّاء (قدم مسطحة) كلاهما مقترنتان بالاضطراب.[4] الأعصاب الحسّية وأعصاب الحس العميق في اليد غالباً ما تكون متضررة، في حين أنّ الأعصاب الألميّة تبقى سليمة. فرط في استعمال اليد أو الطرف المصاب قد يُنشّط بعض الأعراض كالاخدرار والتشنّج والمعص المُؤلم. قد تختلف الأعراض وتقدم المرض. في البعض يمكن أن يتأثر التنفس كما السمع والبصر وعضلات الرقبة والكتف. جَنَف العمود الفقري أمر شائع وقد يكون الحُق (تجويف في عظم الورك) مشوّهاً. يشمل المرض أيضاً مشاكل مَعِدية مِعويّة،[5][6] كذلك صعوبة في المضغ أو البلع أو التكلّم (نتيجةً لضمور الحِبال الصوتية).[7] قد يظهر رُعاش عند تلف العضلات. وقد عُرف أن الحَمل والضوائق الانفعالية الحادة تؤدي إلى تفاقم المرض. على مرضى شاركو-ماري-توث أن يتجنبوا فترات طويلة من عدم الحركة كفترات التعافي من إصابات ثانوية لأن الحركة المحدودة تسارع أعراض المرض بشكل كبير.[8] ألم بسبب تغّير الوضعية، تشوهات هيكليّة، تعب عضلي، معص، كلها شائعة في مرضى شاركو-ماري-توث ويمكن تخفيفها أو معالجتها بالعلاج الطبيعي أو الجراحة أو بالأجهزة التصحيحيّة والمساعِدة. مسكنات الألم قد تزلم أيضاً في حال أن طرق العلاج الأُخرى لم تتخلص من الألم.[9] غالباً ما يكون ألم الاعتلال العصبي أحد أعراض شاركو-ماري-توث لكنه - كبقية الأعراض – يختلف من حالة إلى أخرى، في بعض الحالات يكون الألم من جليل إلى شديد ويؤثر على أنشطة الحياة اليومية. لكنه لا يكون عند جميع المرضى. عندما يتواجد هذا العرض يكون مشابهاً لنفس الألم المصاحب للاعتلالات العصبية المحيطية وأيضاً الألم العصبي التالي للهربِس ومتلازمة الألم الناحي المركب ضمن أمراض أُخرى.[10]
أسباب المرض
ينتج مرض شاركو-ماري-توث من طفرات تؤثر في بروتينات عصبونية. يتم توصيل التَدَفّعات العصبية خلال محوار عصبي محاط بغِمد مياليني. تؤثر معظم الطفرات في شاركو-ماري-توث على الغِمد المياليني، لكن بعضها يؤثر على المحوار العصبي للعصبون. تضاعف منطقة كبير على الذراع القصيرة للصِّبْغِي رقم 17 والتي تحوي الجين PMP22(بروتين الميالين المحيطي 22) هو السبب الأكثر شيوعاً لشاركو-ماري-توث ويعزى إليه 70%-80% من الحالات.توثر بعض الطفرات على جين MFN2والذي يترجم إلى بروتين مُتَقِّدريّ. تحوي الخلايا مجموعات منفصلة من الجينات في نواتها وفي مُتَقِّدراتها. تنتقل المُتَقِّدرات عبر المحوار العصبي في الخلايا العصبية. في بعض أشكال شاركو-ماري-توث، طفرات في بروتين MFN2تؤدي إلى تَعَنْقد المُتَقِّدرات لتصبح غير قادرة على الانتقال عبر المحوار العصبينحو المشبك العصبي. وهذا بدوره يمنع المشبك العصبي من العمل.[11] يقسم شاركو-ماري-توث إلى اعتلالات عصبية أوليّة مزيلة للميَالين (ش.م.ت. 1، ش.م.ت. 3، ش.م.ت 4) واعتلالات عصبية أوليّة مِحوَريّة (ش.م.ت. 2)، مع تداخل متكرر بينها. خلية شُفان هي خلية أخرى لها دور في شاركو-ماري-توث والتي تصنع الغِمْد المَيَاليني من خلال لف غشائها البلازميّ حول المِحْوار العصبي بشكل يشبه السويسرول أو الكعك الملفوف.[12] تعمل كل من العصبونات وخلايا شُفان والأرومَات اللِيفِيَّة معاً لإنتاج عصب فعّال. تتبادل العصبونات وخلايا شُفان الإشارات الجُزيئيّة التي تنظّم البقاء والتمايُز للخلايا. تكون هذه الإشارات معطَّلَة في ش.م.ت. 1. تُكوِّن خلايا شُفان المزيلة للمَيَالين محاور عصبية غير طبيعية البنيان والوظيفة. وقد تؤدي إلى تَنَكُّس مِحْوارِي (تنكسالخلية العصبية نتيجة إصابة أوقطعمحوارها)، أو تتسبب في خلل وظيفي للمحاور. يُمَكِّن الغِمْد المَيَالينيّ الخلايا العصبية من توصيل الإشارات بسرعة. فعند تلف الغِمْد المَيَاليني، تكون إشارات العصب أبطأ، ويمكن قياس ذلك من خلال تخطيط كَهربية العَضل. من ناحية أُخرى، عندما يتضرر المِحْوار فهذا يؤدي إلى جهد فِعْل مُرَكَّبمنخفض.[13]
التشخيص
يمكن تشخيص ش.م.ت. من خلال الأعراض أو من خلال قياس سرعة التدفّع العصبي (دراسات التوصيل العصبي) أو بأخذ خَزعة من العصب أو بفحص الDNA الذي يعطي تشخيصاً نهائياً لكن ليس كل الواصِمْات الجينية لشاركو-ماري-توث معروفة. يُلاحظ ش.م.ت. أول مرة عندما يتَنامى ضعف في أسفل الساق، كاللذي في حالة هبوط القدم؛ أو تشوّهات القدم والذي يشمل التقوّس وأصابع القدم المطرقيّة. لكن هذه العلامات وحدها لا تقود إلى التشخيص. يجب على المرضى أن يُحالوا إلى متخصص في علم الأعصاب أو طب التأهيل. سيطلب متخصص الأعصاب من المرضى أن يمشوا على كُعوبهم لرؤية ضعف العضلات أو أن يقوموا بتحريك جزء من الساق ضد قوة معاكسة. ولتحديد فقد الحسّ سيقوم الطبيب المتخصص بفحص المُنْعَكَس الوترية العميقة كنَفْضة الرُكبة والتي تكون ضعيفة أو غائبة في ش.م.ت. سيسأل الطبيب أيضاً عن سوابق عائلية للإصابة بشاركو-ماري-توث لأنه مرض موروث. مع ملاحظة أن عدم وجود سوابق عائلية لا يستبعد المرض لكنه سيسمح للطبيب باستبعاد مسببات أُخرى للاعتلال العصبي كالسكَّريّ أو التعرض لمواد كيميائية أو أدوية معيّنة.[14] في سنة 2010، كان مرض شاركو-ماري-توث أحد أول الأمراض التي عُرِف فيها المسبب الجيني بدقة من خلال تحليل تسلسل المجموع الجيني (الجينوم) لشخص مصاب. تم ذلك بواسطة علماء يعملون لحساب جمعية شاركو-ماري-توث.[15][16][17] وقد تم تحديد طفرتين في جين SH3TC2 المعروف كمسبب لشاركو-ماري-توث. قام الباحثون بعدها بمقارنة جينوم المريض المصاب بجينوم والدته ووالده وسبعة أخوة وأخوات بعضهم مصاب والبعض الآخر غير مصاب. فوجدوا أن لدى الأب والأم نسخة سليمة ونسخة طافِرة من هذا الجين ولم يعانوا من أعراض أو كانت الأعراض خفيفة. أما الجيل الذي ورث نسختين طافِرتين كان لديهم المرض جليّاً. كانت تكلفة تحليل تسلسل جينوم المريض الأول كاملاً 50000$، لكن قدّر الباحثون أن التكلفة ستصبح 5000 قريباً وستصير شائعة.
تصنيف المرض
ينتج مرض ش.م.ت. من عدة طفرات جينية في عدد من الجينات. واعتماداً على الجين المتأثر يمكن تصنيف المرض إلى أنواع وأنواع فرعية.
المعالجة
بالرغم أنه لا يوجد علاج محدد حالياً إلا أنه قد تم اقتراح حمضالاسكوربيك (فيتامين ج) والذي أظهر بعض الفوائد في نماذج حيوانيّة.[18] وقد تم إجراء تَجربة سريريّة لتحديد مدى فعاليّة إعطاء جرعات عالية من حمض الأسكوربيك لمعالجة البشر المصابين بش.م.ت. النوع 1A.[20] نتائج التجربة على الأطفال أظهرت بأن الجرعات العالية آمنة ولكن لم تصف فعاليّتها.[19] غالباً ما يكون الهدف الأكثر الأهمية لمرضى ش.م.ت. هو المحافظة على الحركة وقوّة العضلات والمرونة. لذلك يُوصى عادةً بالعلاج الطبيعي والنشاط المعتدل لكن يجب تجنّب فَرْط الإجهاد. يجب أن يتم مشاركة المُعالج الطبيعي أو الفيزيائي في تصميم برنامج التمرين والذي يتماشى مع القوّة والمرونة الشخصية للمريض. يمكن أيضاً استخدام تَقويم العظاملتصحيح المشاكل في ش.م.ت. قد يصف المختص في الأطراف الصناعيّة تَقويم الكاحل والقدملعلاج اعتلالات المَشية. تساعد هذه المَقاويم في السيطرة على هبوط القدم واستقرار الكاحل وغالباً ما تُوَفِّر حسّ توازن أفضل للمريض. لبس حذاء ملائم أمر مهم أيضاً لمرضى ش.م.ت. ولكنهم يجدون صعوبة في إيجادها بسبب التقوّس العالي في القدم والأصابع المطرقيّة. وبسبب ضعف الاستقبال حسّي، على مرضى م.ش.ت. أيضاً زيارة طبيب القدم لتقليم الأظافر أو إزالة الجلد السميك الذي يتكوّن في وسادة القدم. ويكون القرار النهائي الذي يمكن اتخاذه هو إجراء عملية جراحيّة من قبل طبيب جراحة العظام أو طبيب القدم ويختار المريض إما أن يثبّت قدمه أو أن يتم تصحيح المشاكل المُترقيّة. تشمل العمليات تقوّيم وتثبيت أصابع القدم وتخفيض التقوّس وأحياناً دمج مفصل الكاحل لتحقيق استقراره. يجب أيضاً الحذر الشديد من السقوط لأن الكسور تحتاج وقتاً أطول لتشفى في حالة مرضى ش.م.ت. كما أن عدم الحركة التابع للكسر يُفاقم من ش.م.ت.[20] قامت جمعية شاركو-ماري-توث باعتبار دواء العلاج الكيميائي فينكرستين Vincristine "كخطر عالي مؤكّد" وأعلنت "قد تم إثبات خطورة فينكرستين ويجب تجنبه من قبل جميع مرضى ش.م.ت. بما فيهم الذين لا يعانون من أيّة أعراض.[21] هناك أيضاً العديد من العمليات التصحيحيّة التي يمكن إجراؤها لتحسين الصحّة الجسديّة. أبلغ بعض المرضى عن استفادتهم من القَنب.
تاريخ المرض


سُمّي المرض نسبةً إلى العلماء الذين وصفوه أول مرة سنة 1886: جان-مارتن شاركو (1825-1893) وتلميذه بيير ماري (1853-1940) في باريس وهنريهواردتوث (1856-1925) في لندن.[22]
الإعلام
تم إنتاج فيلم وثائقي بعنوان "بيرتاديتْ" (Bernadette) والذي يروي قصة آنسة شابة في تصارع المرض. أُصدر الفيلم سنة 2012 ويباع حالياً على الإنترنت ويُعرض مجاناً للمشاهدين الأميريكيين على hulu.com .
أشخاص أصيبوا بهذا المرض
انظر أيضًا
المراجع
- العنوان : Klinická neurologie — ISBN 978-80-7387-389-9
- Krajewski, K. M. (2000). "Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A". Brain 123 (7): 1516–27.doi:10.1093/brain/123.7.1516. ببمد 10869062.
- Physical Medicine and Rehabilitation for Charcot-Marie-Tooth Disease. Medscape. Retrieved March 20th, 2012.
- 3. Le, Tao; Bhushan, Vikas (6 January 2014). First Aid for the USMLE Step 1 2014. McGraw-Hill Education. (ردمك 9780071831420). Retrieved 4 September2014. Typically autosomal dominant inheritance pattern associated with scoliosis and foot deformities (high or flat arches).
- 4. http://www.lindacrabtree.com/cmt/basics/basics_article1.html نسخة محفوظة 2017-04-27 على موقع واي باك مشين.
- 5. Soykan I, McCallum RW (January 1997). "Gastrointestinal involvement in neurologic disorders: Stiff-man and Charcot-Marie-Tooth syndromes". The American Journal of the Medical Sciences 313 (1): 70–3. doi:10.1097/00000441-199701000-00012. ببمد 9001170.
- 6. http://www.ninds.nih.gov/disorders/charcot_marie_tooth/detail_charcot_marie_tooth.htm#265923092 نسخة محفوظة 2016-11-19 على موقع واي باك مشين.
- 7. "Treatment and Management of CMT" (Press release). Charcot-Marie-Tooth Association. October 6, 2010. Retrieved August 26, 2011.
- 8. http://www.patient.co.uk/doctor/charcot-marie-tooth-syndrome-pro نسخة محفوظة 2014-10-17 على موقع واي باك مشين.
- 9. Carter, Gregory T.; Jensen, Mark P.; Galer, Bradley S.; Kraft, George H.; Crabtree, Linda D.; Beardsley, Ruth M.; Abresch, Richard T.; Bird, Thomas D. (1998). "Neuropathic pain in Charcot-Marie-tooth disease". Archives of Physical Medicine and Rehabilitation 79 (12): 1560–4. doi:10.1016/S0003-9993(98)90421-X. ببمد 9862301.
- 10. Baloh, R. H.; Schmidt, R. E.; Pestronk, A.; Milbrandt, J. (2007). "Altered Axonal Mitochondrial Transport in the Pathogenesis of Charcot-Marie-Tooth Disease from Mitofusin 2 Mutations". Journal of Neuroscience 27 (2): 422–30.doi:10.1523/JNEUROSCI.4798-06.2007. ببمد 17215403.
- Berger, Philipp; Young, Peter; Suter, Ueli (2002). "Molecular cell biology of Charcot-Marie-Tooth disease". Neurogenetics 4 (1): 1–15. doi:10.1007/s10048-002-0130-z. ببمد 12030326.
- 14. Yiu, Eppie M.; Burns, Joshua; Ryan, Monique M.; Ouvrier, Robert A. (2008). "Neurophysiologic abnormalities in children with Charcot-Marie-Tooth disease type 1A". Journal of the Peripheral Nervous System 13 (3): 236–241.doi:10.1111/j.1529-8027.2008.00182.x. ببمد 18844790
- 15. http://www.charcot-marie-tooth.org/about_cmt/diagnosis.php[full citation needed] نسخة محفوظة 27 فبراير 2010 على موقع واي باك مشين.
- 16. Wade, Nicholas (2010-03-10). "Disease Cause Is Pinpointed With Genome".New York Times.
- 17. Lupski, James R.; Reid, Jeffrey G.; Gonzaga-Jauregui, Claudia; Rio Deiros, David; Chen, David C.Y.; Nazareth, Lynne; Bainbridge, Matthew; Dinh, Huyen et al. (2010). "Whole-Genome Sequencing in a Patient with Charcot–Marie–Tooth Neuropathy". New England Journal of Medicine 362 (13): 1181–91.doi:10.1056/NEJMoa0908094. ببمد 20220177.
- 18. http://www.cmtausa.org/med_alert.php نسخة محفوظة 2015-05-16 على موقع واي باك مشين.
- 19. Passage, Edith; Norreel, Jean Chrétien; Noack-Fraissignes, Pauline; Sanguedolce, Véronique; Pizant, Josette; Thirion, Xavier; Robaglia-Schlupp, Andrée; Pellissier, Jean François; Fontés, Michel (2004). "Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease". Nature Medicine 10 (4): 396–401. doi:10.1038/nm1023. ببمد 15034573.
- 20. "Neuromuscular Trial/Study". Clinical Trials. Muscular Dystrophy Association. 2007-07-18. Retrieved 2008-05-28.
- 21. Burns, Joshua; Ouvrier, Robert A; Yiu, Eppie M; Joseph, Pathma D; Kornberg, Andrew J; Fahey, Michael C; Ryan, Monique M (2009). "Ascorbic acid for Charcot–Marie–Tooth disease type 1A in children: A randomised, double-blind, placebo-controlled, safety and efficacy trial". Lancet Neurology 8 (6): 537–44.doi:10.1016/S1474-4422(09)70108-5. ببمد 19427269.
- 22. CMT Association: Medical Alert
- 23. http://www.hulu.com/bernadett نسخة محفوظة 1 فبراير 2016 على موقع واي باك مشين.
 بوابة طب
بوابة طب بوابة علوم عصبية
بوابة علوم عصبية